|
|
BMe Kutatói pályázat |
|

Petri László
BMe kutatói pályázat - 2020
![]()
Oláh György Doktori Iskola
BME Vegyészmérnöki és Biomérnöki Kar, Szerves Kémia és Technológia Tanszék
Témavezető: Dr. Keserű György Miklós
Kovalens inhibitorok fejlesztése célpont-specifikus kötőelem-optimálás alkalmazásával
A kutatási téma néhány soros bemutatása
Napjainkra a kovalens inhibitorok fejlesztése jelentős gyógyszerkémiai stratégiává vált. Ezek a vegyületek szerkezetileg két fő részre oszthatók: egy klasszikus gyógyszermolekula-szerű vázszerkezetre és egy, a kovalens kötődés kialakításáért felelős kötőelemre. A megfelelő kötőelem kiválasztása rendkívül fontos a kovalens inhibitorok fejlesztésének korai szakaszában, mivel a célzott aminosavak reakcióképessége és hozzáférhetősége is széles tartományban változhat. PhD-kutatásom során a TTK Gyógyszerkémiai Kutatócsoport kovalens fragmensekkel kapcsolatos kutatási témáiba kapcsolódtam be, ahol többnyire kovalens kötőelemek jellemzésével és célpontspecifikus optimalizálásával foglalkoztam.
A kutatóhely rövid bemutatása
A TTK Gyógyszerkémiai Kutatócsoportot 2013-ban alapította Keserű György Miklós az egykori MTA KKKI Heterociklusos Kémiai Kutatócsoport nyomán. Azóta hazai és nemzetközi együttműködések keretében, kormányzati és iparági kutatási programokban veszünk részt. A kutatócsoport foglalkozik többek között kismolekulás hatóanyagok előállításának kémiai folyamatfejlesztésével, GPCR és kináz típusú fehérjéket célzó fragmensalapú gyógyszerkémiai alapkutatással a számítógépes gyógyszertervezéstől a szintetikus kémiai fejlesztésig bezárólag, valamint kovalens inhibitorok fejlesztésére alkalmas módszerek és molekuláris eszközök kidolgozásával.
A kutatás történetének, tágabb kontextusának bemutatása
A célzott hatóanyag-fejlesztések során az egyik legnagyobb kihívás a hatékonyság és toxicitás közötti helyes egyensúly megtalálása. Egy kovalens inhibitor esetében ennek során a nemkovalens alapváz optimalizálása a toxicitási kockázatok csökkentéséhez, a kötőelem célpontspecifikus optimalizálása pedig a hatékonyság növeléséhez tud hozzájárulni. A kismolekulás kovalens kinázinhibitorok a kovalens inhibitoroknak egy speciális alcsoportját alkotják. A kinázok a sejten belüli szabályozásban kulcsszerepet játszó fehérjék, amelyek változatos biokémiai folyamatokat indíthatnak be szubsztrátfehérjéik foszforilezése útján. Az így szabályozott jelátviteli útvonalak számos megbetegedés esetén játszanak kulcsszerepet, ezek között találunk immun-, kardiovaszkuláris és metabolikus problémákat, valamint daganatos megbetegedéseket is[1]. Ezáltal érthetővé válik, hogy a hatékony kinázinhibitorok tervezése és fejlesztése kiemelkedően fontos gyógyszerkémiai stratégia[2]. A JAK/STAT jelátviteli útvonalban fontos szerepet játszó[3] Janus-kinázok ezen a fehérjék egy alcsoportját képviselik. Az immunrendszer működésében és a vérképzésben betöltött szerepük miatt számos hatóanyag-fejlesztési program célozta már ezt a családot, amelynek tagjai jellemzően valamilyen specifikus citokin- és/vagy növekedésifaktor-receptorokhoz kapcsolódnak. Számos kismolekulás kinázinhibitor létezik, amelyek a JAK-családot célozzák, ezek közül hét kereskedelmi forgalomban is megtalálható. Említésre méltó, hogy e hét gyógyszerkészítmény közül három 2019-ben kapott fogalomba hozatali engedélyt. Megemlítendő továbbá, hogy az aktív centrum erősen konzervált a családhoz tartozó fehérjék körében, így a szelektív gátlás megvalósítása igen nehéz gyógyszerkémiai kihívás. Kovalens inhibitorokkal történő kezelésre azonban lehetőséget nyújthat egy nem konzervált cisztein (C909) aminosav, amely a JAK3 ATP-kötő helyének közelében helyezkedik el[4].
A kutatás célja, a megválaszolandó kérdések
A célzott ciszteinek változatos elhelyezkedése és környezete jelentősen befolyásolhatja azok reakcióképességét és elérhetőségét a protonáltság változásán, a reaktív csoportok térbeli elrendeződésén, az átmeneti állapotok geometriáin, valamint a köztitermékek és a lehetséges reakciótermékek tulajdonságain keresztül[5]. Ezért a megfelelő kovalens kötőelem kiválasztása rendkívül fontos a gyógyszerfejlesztési programok korai szakaszában. Célunk volt egy fragmensalapú kötőelem-optimalizálási módszert fejleszteni (1. ábra).
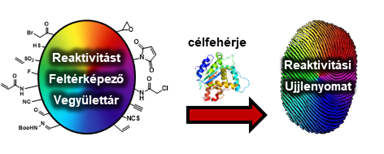
1. ábra: Kötőelem-optimalizálási módszer speciális kovalensfragmens-vegyülettár alkalmazásával.
Noha számos összehasonlító tanulmányt publikáltak már a kötőelemek reaktivitásának témájában[6], az azonos nemkovalens vázra felszerelt kötőelemeket tartalmazó kovalensfragmens-vegyülettár egyedülálló lehetőséget kínál a kötőelem-optimalizálás kovalens gyógyszerfejlesztési folyamatba történő hatékony integrálására. Célunk volt továbbá az is, hogy új kötőelem-optimalizálási módszerünk alkalmazhatóságát demonstráljuk prospektív és retrospektív módon is, gyógyszerkémiai szempontból releváns célpontokon, például kináz fehérjéken.
Módszerek
Annak érdekében, hogy a különböző kötőelemek reaktivitását a hozzájuk kapcsolódó nemkovalens alapváztól függetlenül is jellemezni tudjuk, összeállítottunk egy cisztein-reaktivitást feltérképező speciális kovalensfragmens-vegyülettárat. Az alkalmazott vegyületekben a vizsgált kötőelemek minden esetben egy 3,5-bisz(trifluorometil)fenil- csoporthoz kapcsolódnak (1-28, 2. ábra).
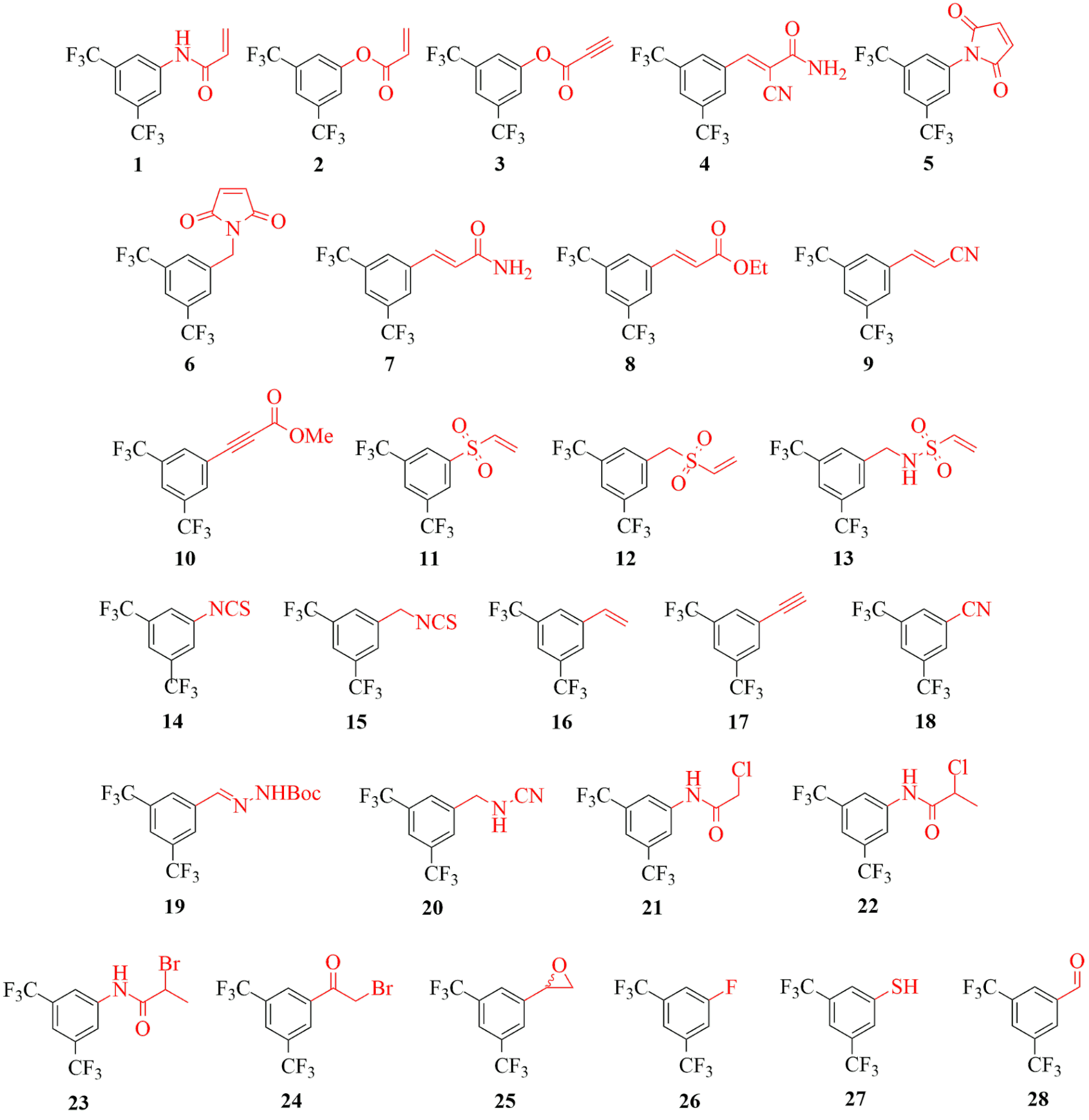
2. ábra: Kovalens fragmensek (1-28) változatos elektrofil kötőelemekkel felszerelve.
A kovalens fragmenseket elméleti és kísérleti reaktivitási paraméterekkel jellemeztük, továbbá vizsgáltuk a vegyületek stabilitását és aminosav-szelektivitását is[7]. A lokális elektrofilicitási indexek alapján megállapítottuk, hogy a vegyülettár a gyengétől az erős elektrofil jellegig széles skálát fed le, majd ugyanezt a konklúziót szolgáltatta a GSH-alapú reaktivitási vizsgálat is. Mivel a célzott ciszteinek reaktivitása maga is jelentősen változhat az adott aminosavra jellemző kémiai környezet függvényében, egy széles reaktivitási ablakot lefedő vegyülettár alkalmas lehet a legtöbb lehetséges célpont reaktivitásának feltérképezésére. Végül azt is megmutattuk, hogy az általunk vizsgált vegyületek minden esetben megfelelő stabilitással rendelkeznek és cisztein-szelektíven reagálnak. Miután jellemeztük a speciális fragmensvegyülettár elemeit reaktivitási, stabilitási és szelektivitási szempontok szerint, a vegyületeket fehérjecélpontokkal szemben is megvizsgáltuk. Ehhez a biológiai szerepek és szerkezeti komplexitás széles skálájáról választottunk reprezentatív fehérjéket (MurA, MAO-A, MAO-B, HDAC8, KRas-G12C és immunoproteaszóma). Mind az egyes célpontok esetében mért egyedi eredmények értékelése, mind a különböző célpontok közötti különbségekre fókuszáló összehasonlító elemzés fontos támpont lehet kovalens inhibitorok tervezése során, és segíthet az adott célpontra leginkább megfelelő kötőelem kiválasztásában. Ezután a vegyülettár alkalmazhatóságának validálása érdekében különböző kinázokon végzett vizsgálatok alapján kifejlesztettünk egy reaktivitásalapú ujjlenyomatprofilt, amelyet azután kovalens JAK3-gátlószerek fejlesztése során használtunk fel.
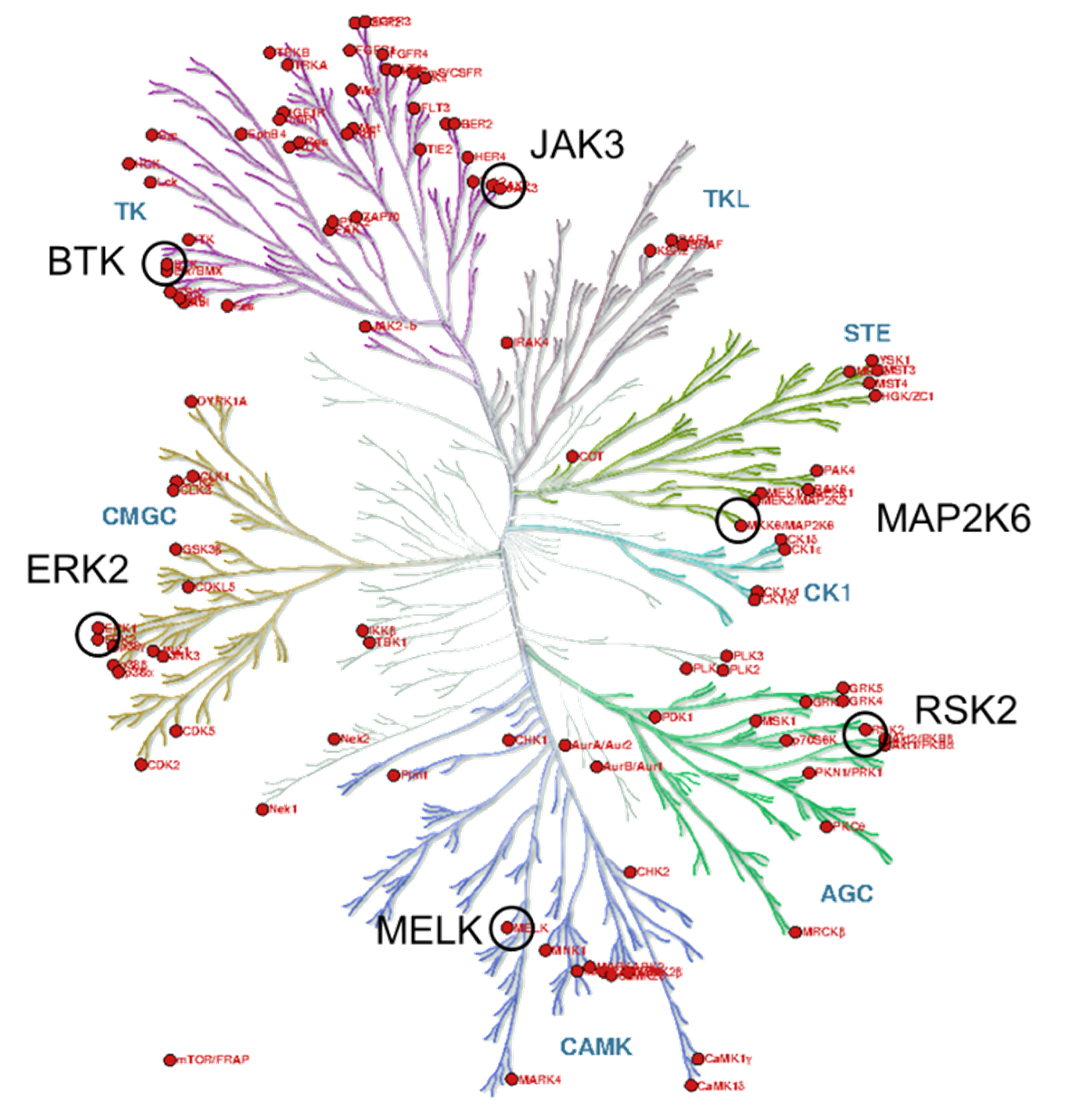
3. ábra: Filogenetikus kinázfa.
A kinázcélpontokat a filogenetikus kinázfa terápiás szempontból releváns és kovalensen már célzott tagjai közül választottuk (3. ábra). Figyelembe véve a ciszteincsoportok elérhetőségét[8], mindegyik fő evolúciós ágról egy-egy kinázt választottunk. Végül a kovalensfragmens-vegyülettár felhasználásával hat kinázra (BTK, ERK2, MELK, RSK2, MAP2K6 és JAK3) határoztuk meg az aktivitási profilt (4. ábra). A lehetséges JAK3-inhibitorokhoz (29-34) a reaktivitásalapú ujjlenyomat alapján választott aktív kötőelemek mellett negatív kontrollként további két inaktív kötőelemet (35, 36) és a nemkovalens acetilcsoportot (37) választottuk ki, hogy megvizsgáljuk az alapváz gátló hatását kovalens kötést kialakító kötőelem nélkül is.
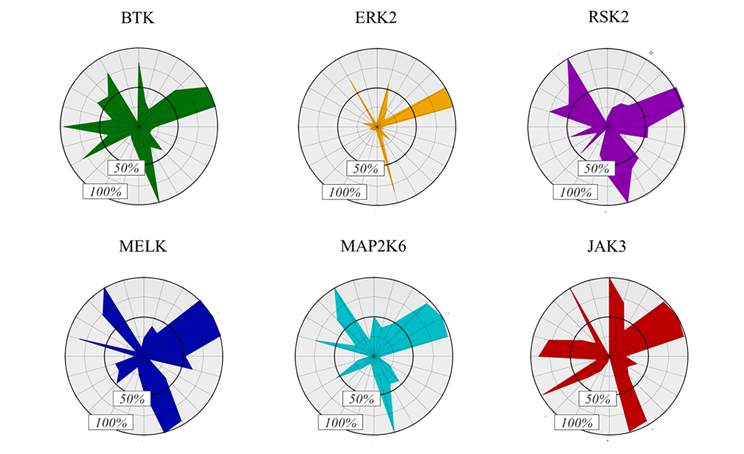
4. ábra: Reaktivitásalapú ujjlenyomatok radardiagramként ábrázolva
A tervezett JAK3-inhibitorok esetében a 4-fenil-pirrolopirimidint választottuk nemkovalens alapvázként. Ez az szerkezet egy jól ismert hinge-kötő motívum, amellyel számos JAK-inhibitor esetében találkozhatunk[9], és amely szintetikusan megvalósítható lehetőséget kínál a kötőelemek optimalizálására. Ezen túlmenően azt is kimutatták, hogy az akrilamid-kötőelemmel felszerelt 4-fenil-pirrolopirimidin kovalensen kötődik a JAK3 ATP-kötő centrumához közeli C909 aminosavhoz[10]. Végül kilenc vegyületet szintetizáltunk, beleértve a potenciális kovalens JAK3-inhibitorokat és a negatív kontrollokat.
Eddigi eredmények
Láthatjuk, hogy néhány kötőelem jól gátolta az összes vizsgált fehérje működését (5. ábra), ezek voltak a maleimidek (5, 6), az akrilvegyületek (2, 7), a vinilszulfonok (11, 12) és az izotiocianát (15). Ezzel szemben volt néhány vegyület, amely egyik vizsgált célponton sem fejtett ki gátló hatást: a sztirol (16), az acetilén (17), a hidrazon (19), a fluorbenzol (26) és az aldehid (28). Kiemelendő, hogy a széles körben alkalmazott akrilamid-kötőelem (1) csak a MurA esetében volt aktív, míg a többi 5 célpont esetében inaktív maradt. Ez már önmagában is jól mutatja, hogy a kovalens inhibitorok fejlesztése során érdemes a kötőelem-kemotípusok szélesebb skálájából kiindulni, hogy optimális reaktivitást és szelektivitást tudjunk ezáltal biztosítani. További bizonyítéka ennek, hogy jól megválasztott kötőelemmel akár MAO-A és MAO-B enzimaltípusok közötti szelektivitást is elérhetünk, ami ebben a esetben nagyon fontos gyógyszerkémiai cél is egyben, hiszen így elkerülhetjük az igen veszélyes „sajtreakció” típusú mellékhatást[11]. A vegyülettárban sikerült azonosítanunk ígéretes MAO-A- és MAO-B-inhibitorokat is. Ezek az eredmények tehát összességében kihangsúlyozzák a kötőelem kemotípusának jelentőségét, hiszen látható, hogy a kötőelem lecserélésével (mindeközben a nemkovalens alapváz változatlanul tartásával) MAO-A-szelektív gátlószert egyszerűen és hatékonyan alakíthatunk át MAO-B-szelektívvé.
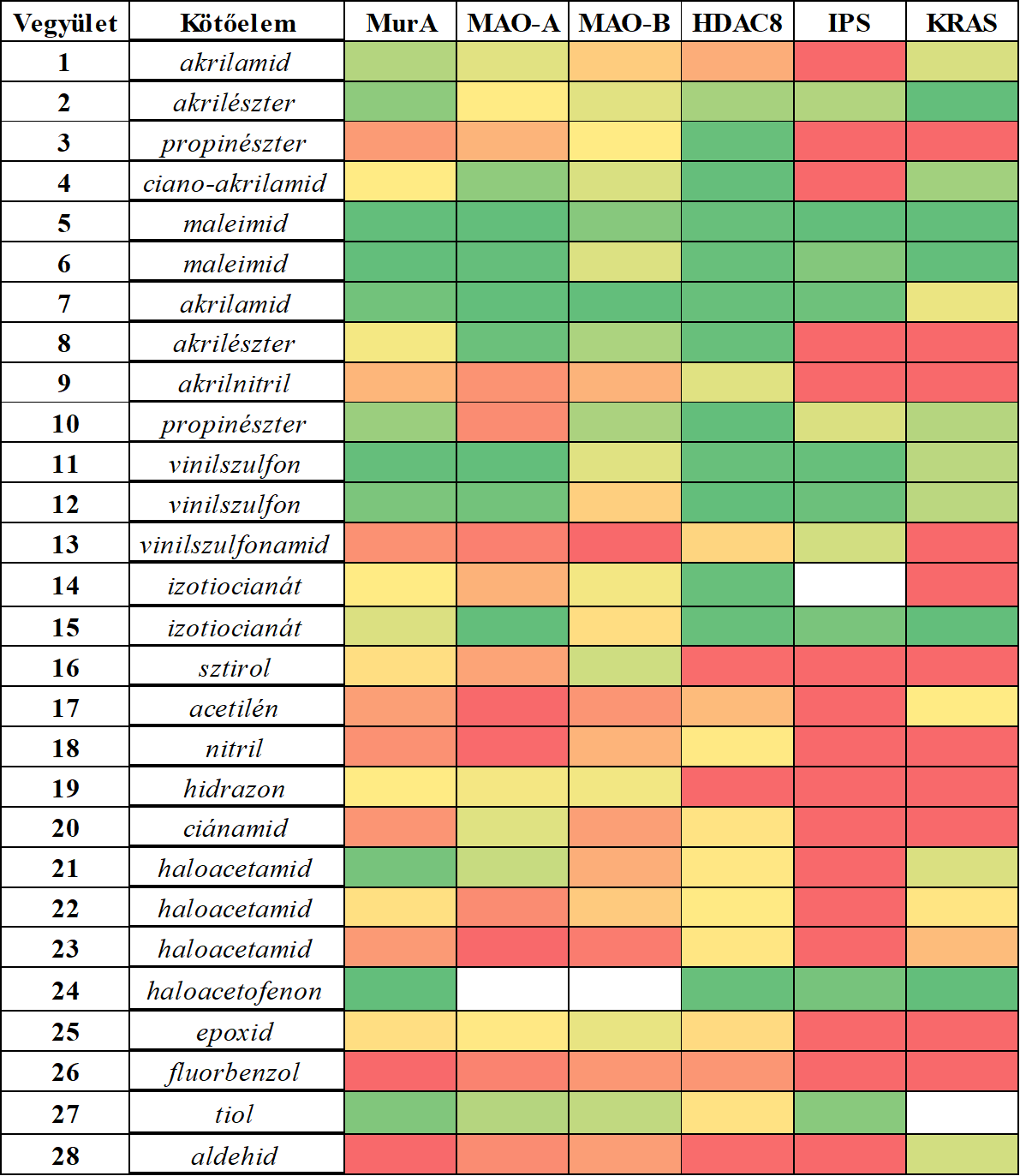
5. ábra: A MurA, MAO-A, MAO-B, HDAC8, immunoproteaszóma (IPS) és KRAS-G12C célfehérjék reaktivitási profiljának összehasonlító elemzése. A hőtérkép színezése az aktivitással van összhangban, a piros színnel jelölt inaktívoktól a zöld színű aktívokig.
Néhány esetben izolált találatok is előfordulhatnak, ami azt jelzi, hogy a kötőelemek egy része egyértelműen egy adott célpontot részesített előnyben. Az akrilnitril (9), fenil-izotiocianát (14), ciánamid (20) és metil-haloacetamidok (22, 23) csak a HDAC8-on voltak aktívak, ami valószínűleg ahhoz köthető, hogy ez a fehérje nagyon reaktív ciszteinnel rendelkezik. Ezeken kívül az akrilamid (1) és a haloacetamid (21) a MurA-t részesítette előnyben a többi 5 célponthoz képest. Összességében sikerült bebizonyítanunk, hogy ez a kísérleti kötőelem-optimalizálási módszer jól alkalmazható a lehetséges gyógyszerkémiai célpontok széles skáláján. A MAO-A esetében ez a vizsgálat emellett egy új kovalens gátlási mechanizmust tárt fel a nemkatalitikus C321 és C323 ciszteinekhez történő kötődéssel. Eredményeink alátámasztják azt a koncepciót, miszerint a különböző célpontokra nem alkalmazható egy univerzális kötőelem. Sőt, a kovalens inhibitorok fejlesztése során nem csak a nemkovalens kölcsönhatások optimalizálása, de a kötőelem- kemotípusok racionális megválasztása is különösen fontos és szükséges lépés. Ezután megvizsgáltuk a kinázaktivitási ujjlenyomatok alapján tervezett vegyületek JAK3-mal szembeni aktivitását (29-37, 1. táblázat).
|
Vegyület |
Kötőelem |
JAK3 |
BTK |
ERK2 |
RSK2 |
MELK |
MAP2K6 |
|
29 |
akrilamid |
< 1 nM |
152 nM |
> 1000 nM |
> 1000 nM |
213 nM |
> 1000 nM |
|
30 |
haloacetamid |
< 1 nM |
61.7 nM |
> 1000 nM |
> 1000 nM |
91 nM |
> 1000 nM |
|
31 |
cianoakrilamid |
55.9 nM |
2940 nM |
> 1000 nM |
399 nM |
25.5 nM |
281 nM |
|
32 |
haloacetamid |
10.6 nM |
342 nM |
> 1000 nM |
> 1000 nM |
216 nM |
> 1000 nM |
|
33 |
maleimid |
26.8 nM |
768 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
|
34 |
tiol |
> 1000 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
|
35 |
aldehid |
379 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
|
36 |
haloacetamid |
140 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
165 nM |
> 1000 nM |
|
37 |
acetamid |
832 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
222 nM |
> 1000 nM |
1. táblázat. JAK3 IC50 értékek az előállított vegyületek esetében (29-37).
Az eredmények alapján sikeresen azonosítottunk irreverzibilis JAK3-inhibitorokat (29, 30) retrospektív módon[12], valamint további vegyületeket (31-33) prospektív módon. Ezek a vegyületek alacsony nanomólos aktivitású gátló hatást fejtettek ki, ezzel alátámasztva az alkalmazott kötőelem-optimalizálási módszer sikerességét. Ezenkívül megállapítottuk, hogy a megfelelő alapváz megválasztásával figyelemre méltó szelektivitást érhetünk el más kinázokkal szemben.
Várható impakt, további kutatás
Eredményeink arra utalnak, hogy az elektrofil kötőelem célpontspecifikus optimalizálásával jelentősen javíthatjuk a kovalens inhibitorok aktivitását és specificitását. Specifikus vegyülettárunk tehát hasznos lehet más gyógyszerfejlesztési programok során is a célzott ciszteinek jellemzésére és a kötőelemek optimalizálására. Összességében, a kovalens fragmensek alkalmazása területén elért eredményeink a kovalens inhibitorok fejlesztése során alkalmazott kötőelemek tudatos megválasztása és optimalizálása révén hozzájárulhatnak a fehérjék kovalens jelölését célzó vegyületek hatékonyságának és szelektivitásának javításához, ezáltal új és hatékony kovalens inhibitorok fejlesztéséhez. Módszerünket szeretnénk a továbbiakban prospektív inhibitorfejlesztés során is validálni olyan gyógyszerkémiai célpontokon, amelyeket eddig nem, vagy csak nehezen lehetett megfelelő kötőelemmel ellátott kovalens inhibitorral szelektíven és hatékonyan gátolni.
Saját publikációk, hivatkozások, linkgyűjtemény
Kapcsolódó saját publikációk listája
[S1] László Petri, Attila Egyed, Dávid Bajusz, Tímea Imre, Péter Ábrányi-Balogh, György M. Keserű: An electrophilic warhead library for mapping the reactivity and accessibility of tractable cysteines in protein kinases, Eur. J. Med. Chem. 2020, 207, 112836. IF: 5.572
[S2] László Petri, Péter Ábrányi-Balogh, Tímea Imre, Gyula Pálfy, Damijan Knez, Martina Hrast, Martina Gobec, Izidor Sosič, Kinga Nyíri, Niklas Jänsch, Charlotte Desczyk, Franz-Josef Meyer-Almes, Simona Golic, Luca Giacinto Iacovino, Claudia Binda, Stanislav Gobec, György M. Keserű: Assessment of tractable cysteines by covalent fragments screening, ChemBioChem., 2020, cbic.202000700. IF: 2.576
[S3] Péter Ábrányi-Balogh, László Petri, Tímea Imre, Péter Szijj, Andrea Scarpino, Martina Hrast, Ana Mitrović, Urša Pečar Fonovič, Krisztina Németh, Hélène Barreteau, David I. Roper, Kata Horváti, György G. Ferenczy, Janko Kos, Janez Ilaš, Stanislav Gobec, György M. Keserű: A road map for prioritizing warheads for cysteine targeting covalent inhibitors, Eur. J. Med. Chem., 2018, 160, 94. IF: 4.816
[S4] László Petri, Péter Ábrányi-Balogh, Petra Regina Varga, Tímea Imre, György Miklós Keserű: Comparative reactivity analysis of small-molecule thiol surrogates, Bioorg. Med. Chem., 2020, 28, 115357. IF: 2.802
[S5] László Petri, Péter A. Szijj, Ádám Kelemen, Tímea Imre, Ágnes Gömöry, Maximillian T. W. Lee, Krisztina Hegedűs, Péter Ábrányi-Balogh, Vijay Chudasama, György Miklós Keserű: Cysteine specific bioconjugation with benzyl isothiocyanates, RSC Adv., 2020, 10, 14928. IF: 3.049
[S6] Aaron Keeley, László Petri, Péter Ábrányi-Balogh, György M. Keserű: Covalent fragment libraries in drug discovery, Drug Discov. Today, 2020, 25, 983. IF: 6.880
[S7] Hung Huy Nguyen, Péter Ábrányi-Balogh, László Petri, Attila Mészáros, Kris Pauwels, Guy Vandenbussche, György M. Keserű, Peter Tompa: Targeting an Intrinsically Disordered Protein by Covalent Modification, Methods in Molecular Biology: Intrinsically Disordered Proteins, szerkesztette: Kragelund B., Skriver K. kiadta: Humana, New York, NY, 2020.
[S8] David J. Hamilton, Péter Ábrányi-Balogh, Aaron Keeley, László Petri, Martina Hrast, Tímea Imre, Maikel Wijtmans, Stanislav Gobec, Iwan J. P. de Esch and György Miklós Keserű: Bromo-Cyclobutenaminones as New Covalent UDP-N-Acetylglucosamine Enolpyruvyl Transferase (MurA) InhibitorsPharmaceuticals, 2020, 13, 362. IF: 4.286
[S9] László Petri: Régi-új fegyver: Kovalens kötéssel a rák ellen, Élet és Tudomány, 2018, 27, 838.
[S10] Andrea Scarpino, László Petri, Damijan Knez, Tímea Imre, Péter Ábrányi-Balogh, György G. Ferenczy, Stanislav Gobec, György M. Keserű: WIDOCK: A Warhead-Independent Virtual Screening Protocol for Cysteine Targeted Covalent Inhibitors, benyújtva (J. Comput. Aid. Mol. Des.)
Egyéb saját publikációk listája
[S11] Tamás Szabó, László Petri, Szilveszter Gergely, Péter Huszthy: Synthesis of achiral and new chiral crown ethers containing a triphenylphosphane unit, Arkivoc, 2015, 5, 20. IF: 1.096
[S12] Krisztina Várnai; László Petri, Lajos Nagy: Prospective evaluation of spent sulfuric acid recovery by process simulation, Period. Polytech. Chem., 2020, doi:10.3311/PPch.15679, IF: 1.382
[S13] Krisztina Várnai and László Petri: Városi farmok: A mezőgazdaság magasiskolája, Élet és Tudomány, 2016, 28, 873.
Linkgyűjtemény
fragmens alapú gyógyszerkutatás
Hivatkozások listája
[1] Grant, S. K. Therapeutic Protein Kinase Inhibitors. Cell. Mol. Life Sci. 66, 1163–1177 (2009)
[2] Santos, R. et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017)
[3] a) Darnell, J. E., et al. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–21 (1994). b) Stark, G. R. & Darnell, J. E. The JAK-STAT Pathway at Twenty. Immunity 36, 503–514 (2012). c) Ihle, J. N. Signaling by the cytokine receptor superfamily just another kinase story. Trends Endocrinol. Metab. 5, 137–43 (1994).
[4] a) Telliez, J.-B. et al. Discovery of a JAK3-Selective Inhibitor: Functional Differentiation of JAK3-Selective Inhibition over pan-JAK or JAK1-Selective Inhibition. ACS Chem. Biol. 11, 3442–3451 (2016) b) Goedken, E. R. et al. Tricyclic Covalent Inhibitors Selectively Target Jak3 through an Active Site Thiol. J. Biol. Chem. 290, 4573–4589 (2015). c) Tan, L. et al. Development of Selective Covalent Janus Kinase 3 Inhibitors. J. Med. Chem. 58, 6589–6606 (2015) d) Kempson, J. et al. Discovery of highly potent, selective, covalent inhibitors of JAK3. Bioorg. Med. Chem. Lett. 27, 4622–4625 (2017)
[5] McGregor, L. M., et al. Expanding the Scope of Electrophiles Capable of Targeting K-Ras Oncogenes. Biochemistry 56, 3178–3183 (2017)
[6] a) Krenske, E. H., et al. Kinetics and Thermodynamics of Reversible Thiol Additions to Mono- and Diactivated Michael Acceptors: Implications for the Design of Drugs That Bind Covalently to Cysteines. J. Org. Chem. 81, 11726–11733 (2016) b) Flanagan, M. E. et al. Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors. J. Med. Chem. 57, 10072–10079 (2014) c) Cee, V. J. et al. Systematic Study of the Glutathione (GSH) Reactivity of N -Arylacrylamides: 1. Effects of Aryl Substitution. J. Med. Chem. 58, 9171–9178 (2015)
[7] Abranyi-Balogh, P. et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors, Eur. J. Med. Chem., 160, 94-107 (2018).
[8] Zhao, Z., et al. Determining Cysteines Available for Covalent Inhibition Across the Human Kinome. J. Med. Chem. 60, 2879–2889 (2017)
[9] a) Mesa, R. A., et al. Ruxolitinib. Nat. Rev. Drug Discov. 11, 103–104 (2012) b) Markham, A. Baricitinib: First Global Approval. Drugs 77, 697–704 (2017) c) Sandborn, W. J. et al. Tofacitinib, an Oral Janus Kinase Inhibitor, in Active Ulcerative Colitis. N. Engl. J. Med. 367, 616–624 (2012) d) Gonzales, A. J. et al. Oclacitinib (APOQUEL®) is a novel Janus kinase inhibitor with activity against cytokines involved in allergy. J. Vet. Pharmacol. Ther. 37, 317–324 (2014)
[10] Thorarensen, A. et al. Design of a Janus Kinase 3 (JAK3) Specific Inhibitor 1-((2 S ,5 R )-5-((7 H -Pyrrolo[2,3- d ]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one (PF-06651600) Allowing for the Interrogation of JAK3 Signaling in Humans. J. Med. Chem. 60, 1971–1993 (2017)
[11] Anderson, M. C.; Hasan, F.; McCrodden, J. M.; Tipton, K. F. Monoamine oxidase inhibitors and the cheese effect, Neurochem. Res. 18, 1145-1149 (1993)
[12] He, L.; Shao, M.; Wang, T.; Lan, T.; Zhang, C.; Chen, L. Design, Synthesis, and SAR Study of Highly Potent, Selective, Irreversible Covalent JAK3 Inhibitors. Mol. Divers. 22, 343–358 (2018)