|
|
BMe Research Grant |
|

Petri László
BMe Research Grant - 2020
![]()
George A. Olah Doctoral School of Chemistry and Chemical Technology
BME Faculty of Chemical Technology and Biotechnology, Department of Organic Chemistry and Technology
Supervisor: Dr. Keserű György Miklós
Development of targeted covalent inhibitors applying target-specific warhead optimization
Introducing the research area
Recently, the research of targeted covalent inhibitors (TCIs) has become a remarkable strategy in the field of medicinal chemistry. These compounds can be structurally divided into two main parts: a classical drug-like scaffold and a warhead directly responsible for the action of the covalent binding. The selection of the appropriate warheads is of utmost importance in the early stages of the development of TCIs, as the reactivity and availability of targeted amino acids can also vary over a wide range. During my PhD I joined the covalent fragment related research projects of the Medicinal Chemistry Research Group, where I mostly worked with characterization and target-specific optimization of warheads.
Brief introduction of the research place
Our research group has been established in 2013 by György M. Keserű on the basis of the preceding MTA KKKI Heterocyclic Chemistry Research Group. Since then, we established national and international, government and industry funded research programs. Among other topics, we research for chemical process developments for the production of small molecule drugs and fragment-based approaches in the lead discovery of GPCR and kinase targets, including computational drug design and synthetic chemical development, either. Furthermore, we do research for methods and molecular tools for the development of targeted covalent inhibitors.
History and context of the research
Research teams working on covalent inhibitors must strike a balance between the efficiency and the toxicity. By the rational design of the non-covalent core of the TCI and target-specific tailoring of the warhead moieties, these programs can provide safe and efficient covalent medicines. Covalent small-molecule kinase inhibitors are forming a specific subclass of the present TCIs. The kinases are one of the major classes of signaling proteins that trigger a variety of biochemical processes through the phosphorylation of different targets. Kinase signaling pathways are involved in various diseases, like immune disorders, cardiovascular and metabolic diseases and most importantly a large number of different cancers[1]. Consequently, there has been a high demand for the design and development of kinase inhibitors[2]. A specific subtype, the Janus Kinases (JAKs) is a member of the JAK/STAT signalling pathway[3]. Their crucial role in the immune system and haematopoiesis inspired numerous drug discovery programs targeting these particular kinases, which are typically attached to specific cytokine and/or growth factor receptors. There are several small molecule kinase inhibitors targeting the JAK family, including seven drugs on the market. Noteworthy, three out of these received approval in 2019. It should be noted that the active site is highly conserved in the JAK family, thus selectively achieving ATP-competitive inhibition is challenging. However, a non-conserved cysteine (C909) residue located near the ATP binding site of JAK3 makes this target tractable for covalent inhibitor programs[4].
The research goals, open questions
The different location and surroundings of the targeted cysteine can influence its reactivity and accessibility through changes in the protonation state, the spatial arrangement of the reacting groups, the geometries of the transition states, the intermediates and the product of the covalent bond-forming reaction[5]. Thus, selecting the appropriate warhead is of utmost importance in the early stages of the drug development process. We intended to develop a ligand-based warhead-optimization methodology by screening a diverse set of covalent fragments (Figure 1).
Figure 1 Warhead-optimization method by applying a specific covalent fragment library.
Although several comparative studies have been published on warhead reactivity[6], our library of warheads attached to the same scaffold represents a unique opportunity for the efficient integration of warhead optimization into the covalent drug development process. Our goal was also to demonstrate the applicability of our novel warhead-optimization method both prospectively and retrospectively, to pharmaceutically relevant drug targets, such as kinases.
Methods
In order to investigate the reactivity of different warheads independently from the non-covalent structural moiety, we decided to equip a single scaffold with a number of warheads, composing a reactivity mapping toolbox. Thus, we equipped a non-covalent fragment, particularly the 3,5-bis(trifluoromethyl)phenyl group with a series of different warhead moieties (1-28, Figure 2).
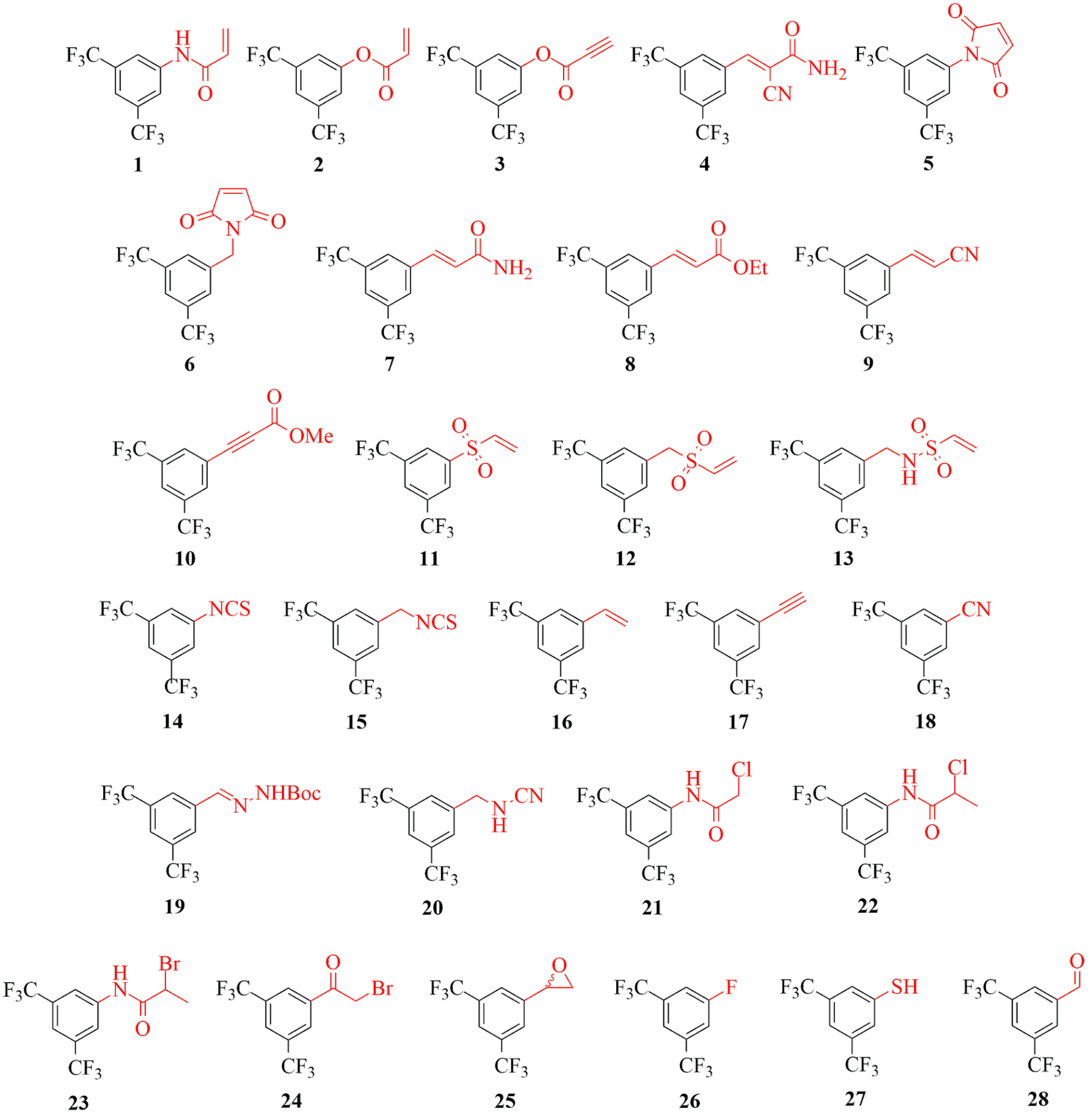
Figure 2 Covalent fragments (1-28) equipped with diverse electrophilic warheads.
The covalent fragments were then characterized by theoretical and experimental reactivity parameters, aqueous stability and amino acid specificity[7]. Assessing local electrophilicity indices we showed that the library covers a wide range of electrophilicity. This has been confirmed by the GSH reactivity data. As the cysteine reactivity is influenced by its individual chemical environment, the broad-spectrum of chemical reactivity is useful for characterizing cysteines with distinct intrinsic reactivities. Furthermore, the fragments showed appropriate aqueous stability for biology testing and the cysteine selectivity was also confirmed. After exploring theoretical and experimental reactivity range of the library members, we investigated different target proteins including a range of enzymes with different functional and structural complexity (MurA, MAO-A, MAO-B, HDAC8, KRas-G12C and immunoproteasome). Both the individual and the comparative analysis of the profiling results might help identify specificity issues in covalent design, and it can suggest the best warheads for each covalent inhibitor program. Next, we intended to validate the applicability of our specific library. Thus, by screening the covalent fragment library against a range of different kinases, we developed a reactivity-based fingerprint, which was then used for warhead selection for specific JAK3 covalent inhibitors.
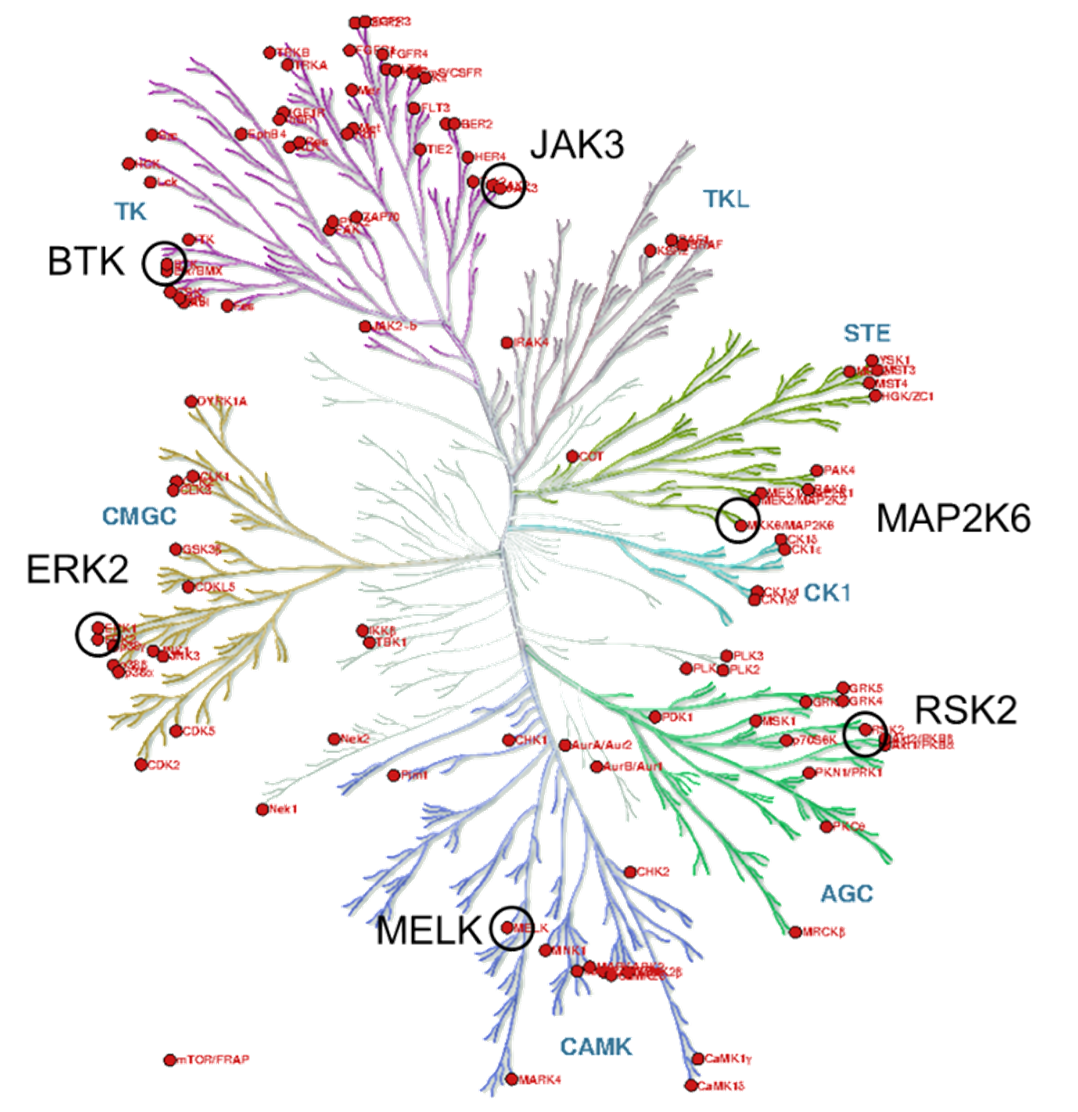
Figure 3 The kinase phylogenetic tree.
The target kinases were selected from the therapeutically significant and already covalently targeted members of the kinase phylogenetic tree (Figure 3). Taking into account the availability of covalently targetable cysteine residues [8], one kinase was selected from each major evolutionary branches; finally, six kinases (BTK, ERK2, MELK, RSK2, MAP2K6 and JAK3) have been included (Figure 4). For the development and investigation of JAK3-inhibitors (29-34) beside the active warheads (based on the warhead reactivity fingerprint) we additionally selected two inactive warheads (35, 36), as negative control, and the non-covalent acetyl group (37) to investigate the inhibitory activity of the binder without a warhead capable of forming a covalent bond.
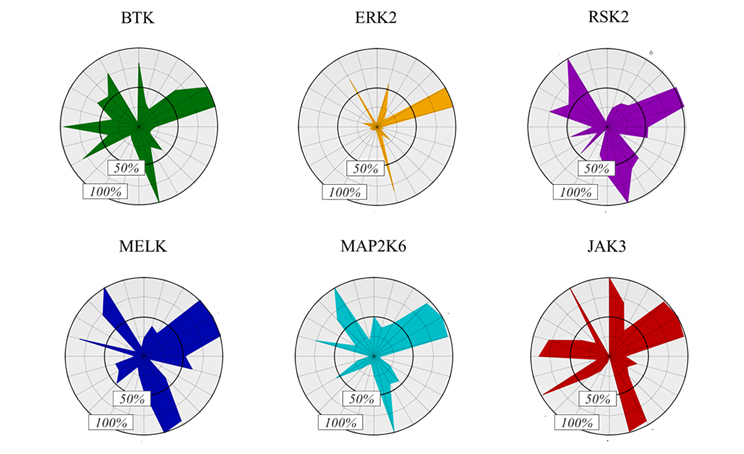
Figure 4 Activity-based fingerprints shown as radar plots.
We selected the 4-phenyl-pyrrolopyrimidine core as the non-covalent scaffold of the designed JAK3 inhibitors. This scaffold is a well-established hinge binder, utilized by many JAK inhibitors [9], providing a synthetically feasible option for warhead optimization. Moreover, the scaffold, armed with an acrylamide warhead was recently shown to bind to the JAK3 ATP-site covalently through C909[10]. Finally, we synthesized nine compounds for JAK3, including the potentially covalent JAK3 binders and the negative controls.
Results
We can see that some warheads showed very promiscuous reactivity inhibiting most of the targeted proteins (Figure 5). These were the maleimides (5, 6), acrylic compounds (2, 7), the vinylsulfones (11, 12) and the isothiocyanate (15). In contrary, there were a few compounds inhibiting none of the investigated targets, such as the styrene (16), the ethinyl (17), the hydrazone (19), the fluorobenzene (26) and the aldehyde (28). Interestingly, we found that the most widely used terminal acrylamide (1) motif was active only for MurA and remained inactive for the other five targets. This again is a strong argument for considering a wider range of warhead chemotypes to gain the optimal reactivity and specificity. This is supported further by the observation, that a good choice of warhead can induce specificity even between MAO-A and MAO-B. Subtype-selectivity issues are critical in the case of MAO inhibition, as non-selective MAO-inhibitors could cause dangerous adverse events, such as the “cheese reaction”[11]. Interestingly, here we obtained promisingly selective inhibitors preferring MAO-A or MAO-B. These results underline the importance of the warhead chemistry, as replacing the warhead on the identical non-covalent scaffold can turn a MAO-A selective inhibitor to just a MAO-B selective one.
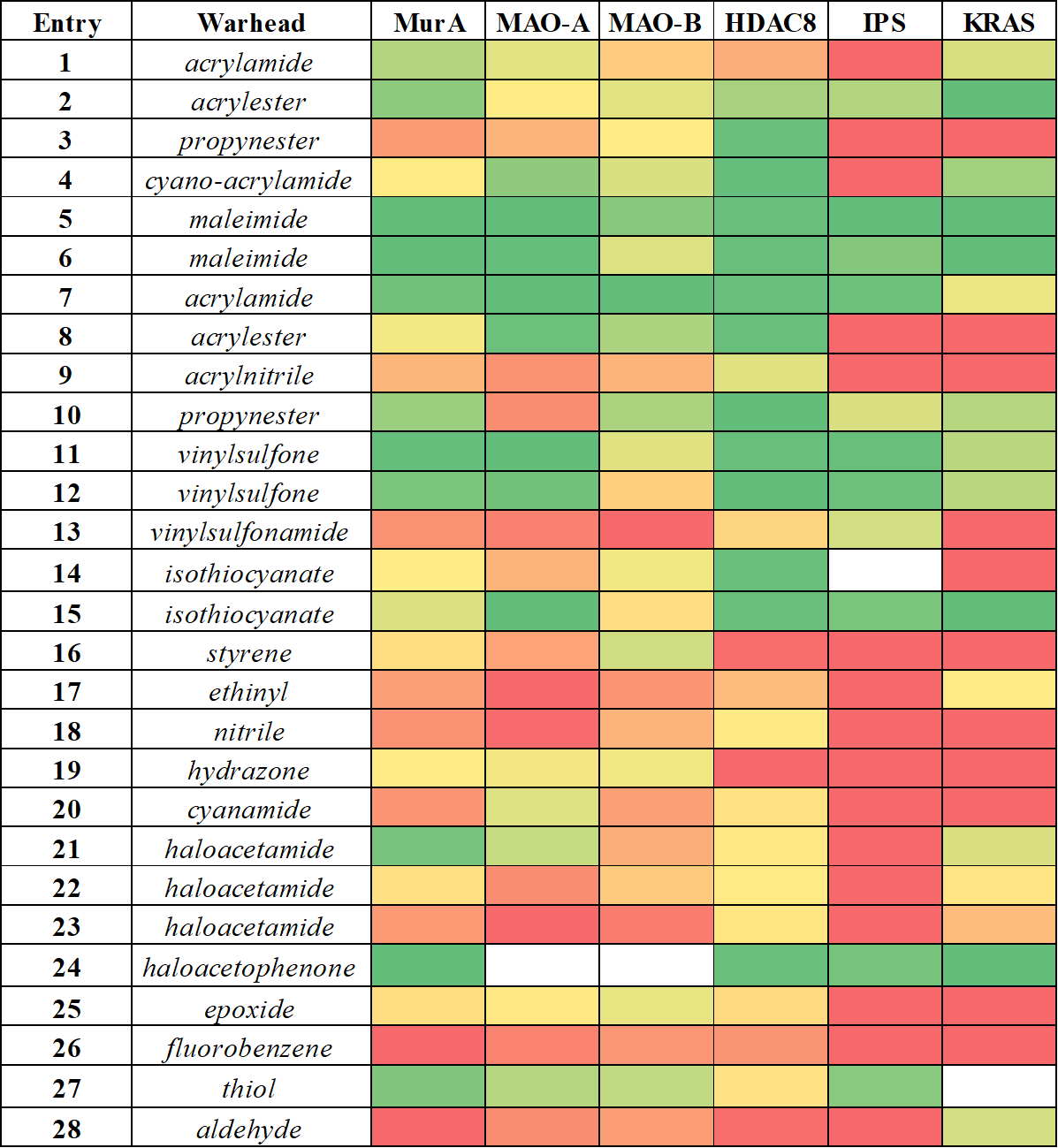
Figure 5 Comparative analysis of the reactivity profile for mapping library obtained for MurA, MAO-A, MAO-B, HDAC8, immunoproteasome (IPS) and KRAS-G12C. Heat map coloring is consistent with activity, ranging from the inactives in red to the actives in green.
There are some isolated hits, indicating that some of the warheads showed clear preference for one particular target. Acrylnitrile (9), phenyl-isothiocyanate (14), cyanamide (20) and methylated haloacetamides (22, 23) was active only on HDAC8, which is most likely related to its high cysteine reactivity. Furthermore, acrylamide (2) and haloacetamide (21) preferred MurA over the other 5 proteins. Altogether, we could show that this methodology is applicable over a wide range of targets as covalent binding was demonstrated with all enzymes. In the case of MAO-A, this analysis also revealed a new covalent mechanism of inhibition by binding to the non-catalytic C321 and C323. Our results support the notion that there is no universal warhead available for different targets. In fact, the required specificity of TCIs necessitates not only the optimized non-covalent interactions, but also the careful selection and tailoring of the warheads. Next we investigated the activity of JAK3 inhibitors (29-37, Table 1), designed by the kinase activity fingerprint.
|
Entry |
Warhead |
JAK3 |
BTK |
ERK2 |
RSK2 |
MELK |
MAP2K6 |
|
29 |
acrylamide |
< 1 nM |
152 nM |
> 1000 nM |
> 1000 nM |
213 nM |
> 1000 nM |
|
30 |
haloacetamide |
< 1 nM |
61.7 nM |
> 1000 nM |
> 1000 nM |
91 nM |
> 1000 nM |
|
31 |
cyanoacrylamide |
55.9 nM |
2940 nM |
> 1000 nM |
399 nM |
25.5 nM |
281 nM |
|
32 |
haloacetamide |
10.6 nM |
342 nM |
> 1000 nM |
> 1000 nM |
216 nM |
> 1000 nM |
|
33 |
maleimide |
26.8 nM |
768 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
|
34 |
thiol |
> 1000 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
|
35 |
aldehyde |
379 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
|
36 |
haloacetamide |
140 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
165 nM |
> 1000 nM |
|
37 |
acetamide |
832 nM |
> 1000 nM |
> 1000 nM |
> 1000 nM |
222 nM |
> 1000 nM |
Table 1 JAK3 IC50 values of the designed compounds (29-37).
The results confirmed that the already described[12] inhibitors (29, 30) could be identified retrospectively, while other compounds (31-33) provided prospective hits for irreversible inhibition of JAK3. These compounds exhibited low nanomolar inhibitory activities, confirming the applicability of the warhead optimization approach we developed. Furthermore, by choosing an appropriate scaffold, we could demonstrate remarkable selectivity against other kinases.
Expected impact and further research
Our results suggest that target-specific optimization of the warhead can significantly improve the activity and the specificity of the TCIs. Our probe library can be useful in other TCI discovery programs for characterizing the targeted cysteines and optimizing the warhead. Overall, the application of covalent fragments can contribute to the proper selection and optimization of warheads in TCI development programs. These efforts can be rationalized by the improvements of efficiency and selectivity, so finally, new and successful targeted covalent inhibitors can be reached. We would like to further validate our target-specific warhead-optimization technique for prospective inhibitor developments on relevant therapeutic targets which, up to date, were difficult or even impossible to selectively and effectively inhibit with targeted covalent inhibitors.
Publications, references, links
List of corresponding own publications
[S1] László Petri, Attila Egyed, Dávid Bajusz, Tímea Imre, Péter Ábrányi-Balogh, György M. Keserű: An electrophilic warhead library for mapping the reactivity and accessibility of tractable cysteines in protein kinases, Eur. J. Med. Chem. 2020, 207, 112836. IF: 5.572
[S2] László Petri, Péter Ábrányi-Balogh, Tímea Imre, Gyula Pálfy, Damijan Knez, Martina Hrast, Martina Gobec, Izidor Sosič, Kinga Nyíri, Niklas Jänsch, Charlotte Desczyk, Franz-Josef Meyer-Almes, Simona Golic, Luca Giacinto Iacovino, Claudia Binda, Stanislav Gobec, György M. Keserű: Assessment of tractable cysteines by covalent fragments screening, ChemBioChem., 2020, cbic.202000700. IF: 2.576
[S3] Péter Ábrányi-Balogh, László Petri, Tímea Imre, Péter Szijj, Andrea Scarpino, Martina Hrast, Ana Mitrović, Urša Pečar Fonovič, Krisztina Németh, Hélène Barreteau, David I. Roper, Kata Horváti, György G. Ferenczy, Janko Kos, Janez Ilaš, Stanislav Gobec, György M. Keserű: A road map for prioritizing warheads for cysteine targeting covalent inhibitors, Eur. J. Med. Chem., 2018, 160, 94. IF: 4.816
[S4] László Petri, Péter Ábrányi-Balogh, Petra Regina Varga, Tímea Imre, György Miklós Keserű: Comparative reactivity analysis of small-molecule thiol surrogates, Bioorg. Med. Chem., 2020, 28, 115357. IF: 2.802
[S5] László Petri, Péter A. Szijj, Ádám Kelemen, Tímea Imre, Ágnes Gömöry, Maximillian T. W. Lee, Krisztina Hegedűs, Péter Ábrányi-Balogh, Vijay Chudasama, György Miklós Keserű: Cysteine specific bioconjugation with benzyl isothiocyanates, RSC Adv., 2020, 10, 14928. IF: 3.049
[S6] Aaron Keeley, László Petri, Péter Ábrányi-Balogh, György M. Keserű: Covalent fragment libraries in drug discovery, Drug Discov. Today, 2020, 25, 983. IF: 6.880
[S7] Hung Huy Nguyen, Péter Ábrányi-Balogh, László Petri, Attila Mészáros, Kris Pauwels, Guy Vandenbussche, György M. Keserű, Peter Tompa: Targeting an Intrinsically Disordered Protein by Covalent Modification, Methods in Molecular Biology: Intrinsically Disordered Proteins, edited: Kragelund B., Skriver K. published: Humana, New York, NY, 2020.
[S8] David J. Hamilton, Péter Ábrányi-Balogh, Aaron Keeley, László Petri, Martina Hrast, Tímea Imre, Maikel Wijtmans, Stanislav Gobec, Iwan J. P. de Esch and György Miklós Keserű: Bromo-Cyclobutenaminones as New Covalent UDP-N-Acetylglucosamine Enolpyruvyl Transferase (MurA) InhibitorsPharmaceuticals, 2020, 13, 362. IF: 4.286
[S9] László Petri: Régi-új fegyver: Kovalens kötéssel a rák ellen, Élet és Tudomány, 2018, 27, 838.
[S10] Andrea Scarpino, László Petri, Damijan Knez, Tímea Imre, Péter Ábrányi-Balogh, György G. Ferenczy, Stanislav Gobec, György M. Keserű: WIDOCK: A Warhead-Independent Virtual Screening Protocol for Cysteine Targeted Covalent Inhibitors, submitted (J. Comput. Aid. Mol. Des.)
List of other own publications
[S11] Tamás Szabó, László Petri, Szilveszter Gergely, Péter Huszthy: Synthesis of achiral and new chiral crown ethers containing a triphenylphosphane unit, Arkivoc, 2015, 5, 20. IF: 1.096
[S12] Krisztina Várnai; László Petri, Lajos Nagy: Prospective evaluation of spent sulfuric acid recovery by process simulation, Period. Polytech. Chem., 2020, doi:10.3311/PPch.15679, IF: 1.382
[S13] Krisztina Várnai and László Petri: Városi farmok: A mezőgazdaság magasiskolája, Élet és Tudomány, 2016, 28, 873.
Table of links
covalent bondfragment-based drug discovery
List of references
[1] Grant, S. K. Therapeutic Protein Kinase Inhibitors. Cell. Mol. Life Sci. 66, 1163–1177 (2009)
[2] Santos, R. et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017)
[3] a) Darnell, J. E., et al. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–21 (1994). b) Stark, G. R. & Darnell, J. E. The JAK-STAT Pathway at Twenty. Immunity 36, 503–514 (2012). c) Ihle, J. N. Signaling by the cytokine receptor superfamily just another kinase story. Trends Endocrinol. Metab. 5, 137–43 (1994).
[4] a) Telliez, J.-B. et al. Discovery of a JAK3-Selective Inhibitor: Functional Differentiation of JAK3-Selective Inhibition over pan-JAK or JAK1-Selective Inhibition. ACS Chem. Biol. 11, 3442–3451 (2016) b) Goedken, E. R. et al. Tricyclic Covalent Inhibitors Selectively Target Jak3 through an Active Site Thiol. J. Biol. Chem. 290, 4573–4589 (2015). c) Tan, L. et al. Development of Selective Covalent Janus Kinase 3 Inhibitors. J. Med. Chem. 58, 6589–6606 (2015) d) Kempson, J. et al. Discovery of highly potent, selective, covalent inhibitors of JAK3. Bioorg. Med. Chem. Lett. 27, 4622–4625 (2017)
[5] McGregor, L. M., et al. Expanding the Scope of Electrophiles Capable of Targeting K-Ras Oncogenes. Biochemistry 56, 3178–3183 (2017)
[6] a) Krenske, E. H., et al. Kinetics and Thermodynamics of Reversible Thiol Additions to Mono- and Diactivated Michael Acceptors: Implications for the Design of Drugs That Bind Covalently to Cysteines. J. Org. Chem. 81, 11726–11733 (2016) b) Flanagan, M. E. et al. Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors. J. Med. Chem. 57, 10072–10079 (2014) c) Cee, V. J. et al. Systematic Study of the Glutathione (GSH) Reactivity of N -Arylacrylamides: 1. Effects of Aryl Substitution. J. Med. Chem. 58, 9171–9178 (2015)
[7] Abranyi-Balogh, P. et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors, Eur. J. Med. Chem., 160, 94-107 (2018).
[8] Zhao, Z., et al. Determining Cysteines Available for Covalent Inhibition Across the Human Kinome. J. Med. Chem. 60, 2879–2889 (2017)
[9] a) Mesa, R. A., et al. Ruxolitinib. Nat. Rev. Drug Discov. 11, 103–104 (2012) b) Markham, A. Baricitinib: First Global Approval. Drugs 77, 697–704 (2017) c) Sandborn, W. J. et al. Tofacitinib, an Oral Janus Kinase Inhibitor, in Active Ulcerative Colitis. N. Engl. J. Med. 367, 616–624 (2012) d) Gonzales, A. J. et al. Oclacitinib (APOQUEL®) is a novel Janus kinase inhibitor with activity against cytokines involved in allergy. J. Vet. Pharmacol. Ther. 37, 317–324 (2014)
[10] Thorarensen, A. et al. Design of a Janus Kinase 3 (JAK3) Specific Inhibitor 1-((2 S ,5 R )-5-((7 H -Pyrrolo[2,3- d ]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one (PF-06651600) Allowing for the Interrogation of JAK3 Signaling in Humans. J. Med. Chem. 60, 1971–1993 (2017)
[11] Anderson, M. C.; Hasan, F.; McCrodden, J. M.; Tipton, K. F. Monoamine oxidase inhibitors and the cheese effect, Neurochem. Res. 18, 1145-1149 (1993)
[12] He, L.; Shao, M.; Wang, T.; Lan, T.; Zhang, C.; Chen, L. Design, Synthesis, and SAR Study of Highly Potent, Selective, Irreversible Covalent JAK3 Inhibitors. Mol. Divers. 22, 343–358 (2018)