|
|
BMe Research Grant |
|

Dargó Gergő
BMe Research Grant - 2020
![]()
George A. Olah Doctoral School of Chemistry and Chemical Technology
BME VBK, Gedeon Richter Plc.
Supervisor: Dr. Balogh György Tibor
Novel techniques for physicochemical profiling of drug-macromolecule interactions
Introducing the research area
Investigation of the interactions between drugs and macromolecules has a pivotal role in certain phases of the drug discovery process. There is a fundamental need for the application of complexing additives (e.g., cyclodextrins [1], proteins, liposomes) to promote absorption of poorly water-soluble drugs, increasing their bioavailability [2]. Furthermore, complexation greatly influences the distribution of the active pharmaceutical ingredients (API) in the human body and their delivery to the site of action (e.g., by binding to serum proteins [3]). To this avail, the development of up-to-date analytical methods is of utmost importance.
Brief introduction of the research place
Since 2005 at Gedeon Richter Plc., and since 2019 September in the Biomimetic Technologies Research Group of the Department of Chemical and Environmental Process Engineering, the physicochemical and pharmacokinetic profiling of drug candidates (solubility, acid/base properties, lipophilicity, permeability) is carried out by in vitro methods.
History and context of the research
Adequate analytical methods are indispensable to study interactions between drugs and macromolecules which are influencing the stability of the complex, and the bioavailability of the drugs. These techniques can be generally classified into two groups: separation based (e.g., affinity capillary electrophoresis, chromatographic techniques, equilibrium dialysis, mass spectrometry) and non-separation based techniques (e.g., phase-solubility, spectrophotometry, potentiometry, NMR spectroscopy, conductometry, hydrolysis kinetics, permeability measurements) [4,5]. The former separates the free and complexed drug and quantifies their concentrations, while the latter group of methods monitors the change in specific physicochemical properties of the drug or macromolecule.
The research goals, open questions
Since the stability of the formed complex has a direct influence on the pharmacokinetic and pharmacodynamic behavior of drugs, the knowledge of this parameter is also fundamental in the early stage of drug discovery. My aim was to develop novel, robust, and cost-effective medium- or high-throughput methods to study the interactions present between drugs and macromolecules.
During my research I studied the following topics regarding drug–macromolecule interactions:
1) Determination of stability constants of inclusion complexes of cyclodextrins (CD) and drugs possessing ionization center(s) based on the changes in partition coefficients of the drugs, using potentiometric titration. Comparison of the measured stability constants with ones determined by conventional methods.
2) Development of an in vitro, non-cellular model to predict corneal drug permeability of eye drops and ophthalmic formulations using the PAMPA (Parallel Artificial Membrane Permeability Assay) technique [6]. Refinement of the corneal-PAMPA model to investigate the effect of complexing agents on corneal permeability. Further testing of the model on a large number of drugs, comparison of the results with permeability data measured with a cellular model (Caco-2) and physicochemical parameters to verify the orthogonality of the method. Development of a computer-based (in silico) model for rapid prediction of corneal permeability.
3) Studying the applicability of UV-pH titration as an alternative to determine human serum albumin binding of drugs, by monitoring changes in proton dissociation constants (pKa). Comparison of the results with literature data and experimentally determined values using orthogonal techniques (liquid chromatography (HPLC) [7], rapid equilibrium dialysis (RED) [8]) and in silico molecular modeling.
4) Investigation of the permeability and dissolution of HSA nanoparticles containing a non-steroidal anti-inflammatory drug (meloxicam) developed for intranasal administration for central nervous system (CNS) targeted therapy.
Methods
1) To determine the stability constants of CD–drug complexes potentiometric titration was used. Partition coefficients (logP) and apparent partition coefficients (logPapp) of two acidic drugs (diclofenac, ketoprofen) and two basic drugs (bupropion, promethazine) were determined at various (2-hydroxypropyl)-ß-CD (HPBCD) concentrations. Due to the presence of octanol–HPBCD complexes in the system, a correction to the phase ratio was also included in the model.
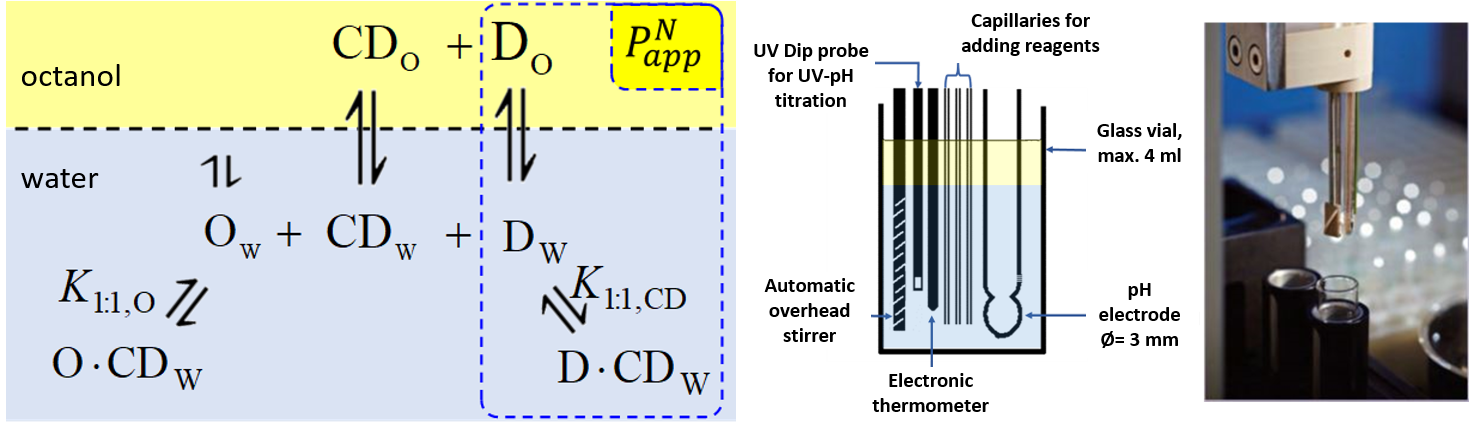
Figure 1 Equilibria between drug (D), cyclodextrin (CD), and octanol (O) molecules in the octanol–water biphasic system and titration cell of the SiriusT3 instrument.
2) During the measurement of APIs of eye drops and ophthalmic formulations, measurement conditions (lipid composition of the membrane, presence of cosolvent, buffer) of the sandwich-like, 96-well based PAMPA system were optimized for the measurement of corneal permeability, based on ex vivo rabbit corneal permeability values. After incubation of the plates at 35 °C for 4 hours, the concentration of the APIs was determined in the donor and acceptor wells by chromatographic methods (HPLC-DAD).
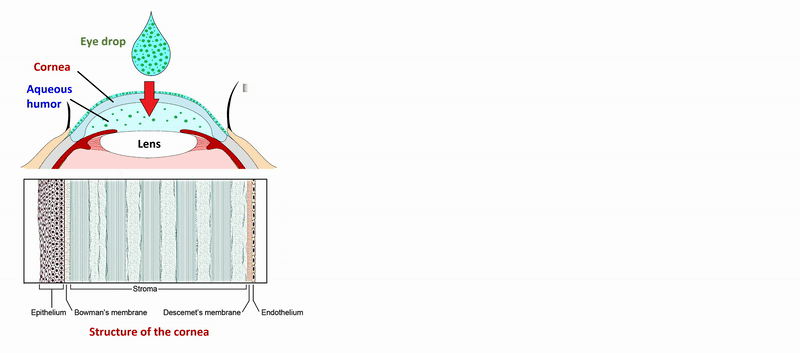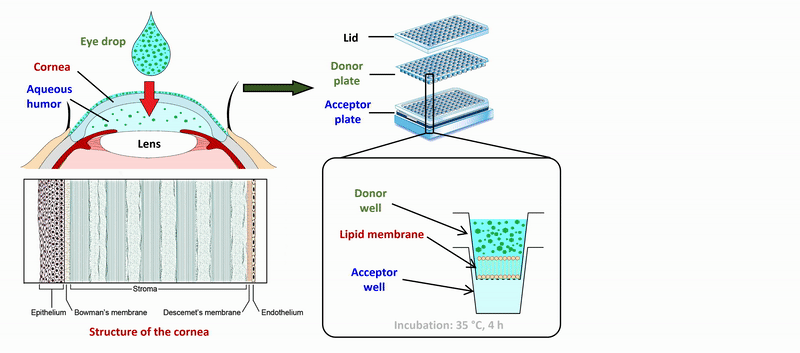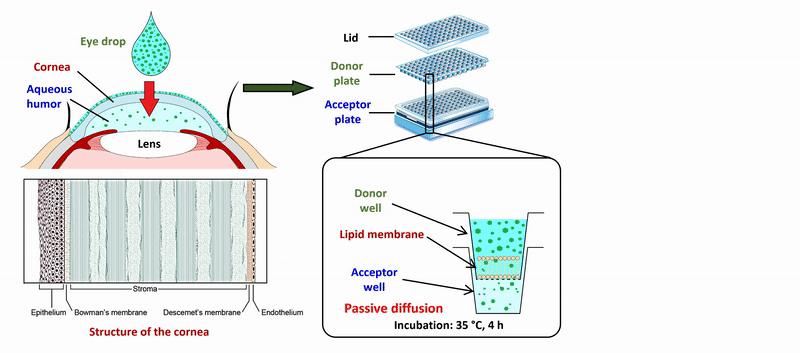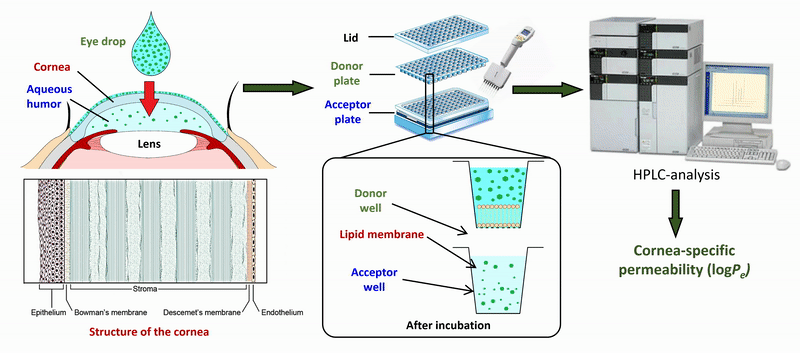
Figure 2 Flow chart of the determination of corneal permeability using the PAMPA system.
The optimized model was also tested in the case of eye drops and ophthalmic formulations. Based on the permeability data measured for a large number of drugs, we also developed an in silico model using a PLS-based (partial least squares regression) method using physicochemical descriptors and molecular fingerprints.
3) For the determination of human serum albumin (HSA) binding of drugs, we used the UV-pH titration technique based on previous experience with CD–drug complexes [FP9]. The technique provides a fast and robust way to measure pKa values of molecules with a pH-dependent absorbance spectrum. The presence of HSA during titration may change the pH-dependent spectrum and therefore the pKa values of a molecule. Based on the extent of the pKa shift, we can estimate the HSA binding affinity of the drug. We compared our results to experimental values determined by conventional methods: a chromatographic analysis using an immobilized HSA column and rapid equilibrium dialysis (RED). In some cases, in silico molecular docking was also carried out to support our results.
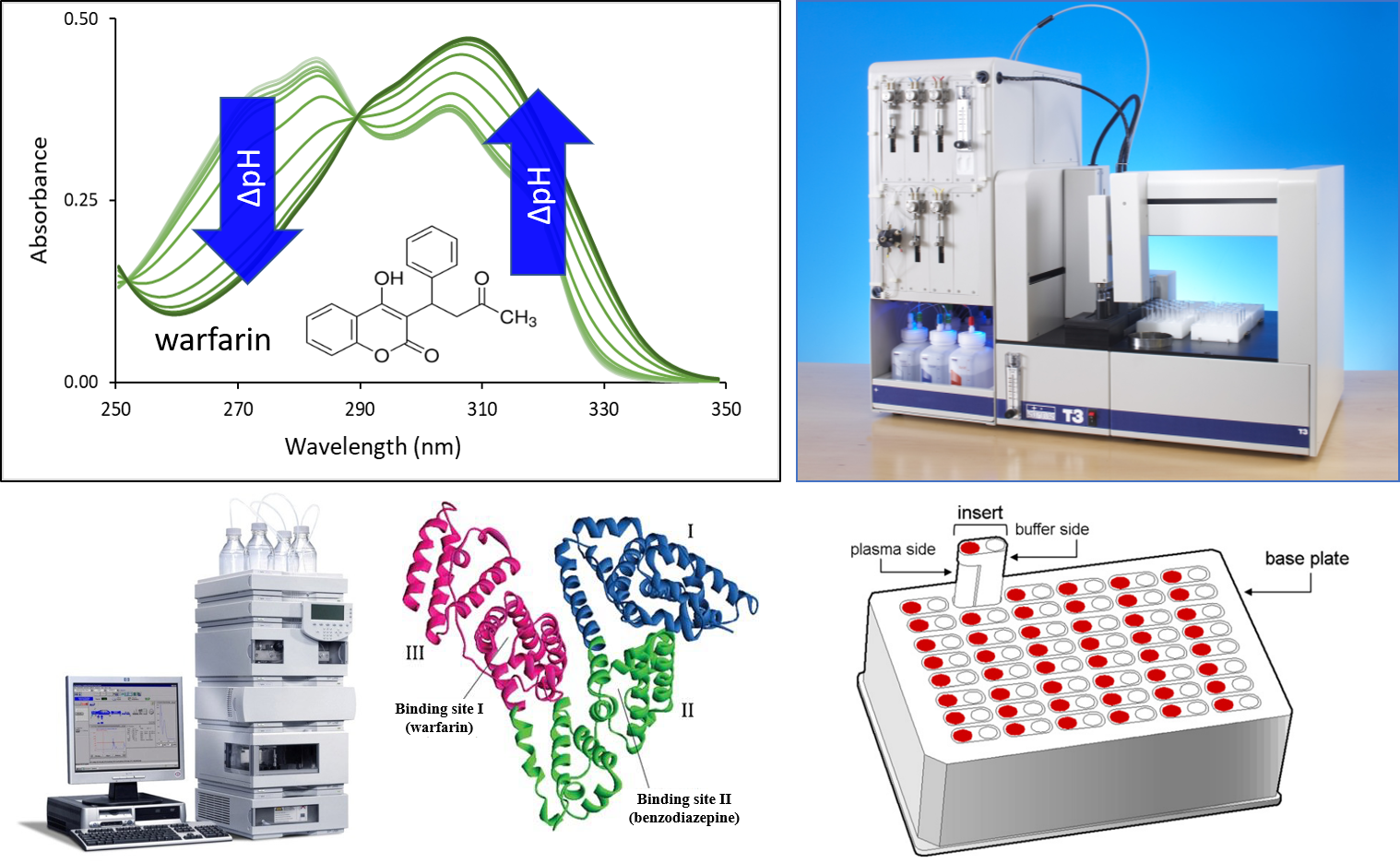
Figure 3 Techniques for the determination of HSA binding: UV-pH titration (above), HPLC measurement using immobilized HSA column and rapid equilibrium dialysis (RED) (below).
4) In cooperation with the University of Szeged, we participated in the physicochemical characterization of HSA nanoparticles prepared by their research group. The dissolution profile was measured at pH 5.6 (nasal mucosa), using the semipermeable membrane system of the RED plate. The permeability measurements were carried out using the PAMPA system (donor/acceptor pH 5.6/7.4, membrane: 2.7 w/V% phosphatidylcholine + 1.3 w/V% cholesterol dissolved in dodecane, incubation: at 37 °C, 4 hours). The MEL concentration was determined by HPLC-DAD in both cases.
Results
1) In the case of potentiometric titrations, the collected data points are transformed into Bjerrum plots, which show well the differences between proton dissociation constants (pKa) measured in water, octanol–water, and analogous systems containing HPBCD. Afterward, the Henderson-Hasselbalch equation can be used to calculate partition coefficients and apparent partition coefficients of the drug in the presence and absence of HPBCD (logPapp és logP), based on pKa and poKa values measured in the water, octanol–water systems (Fig. 4).
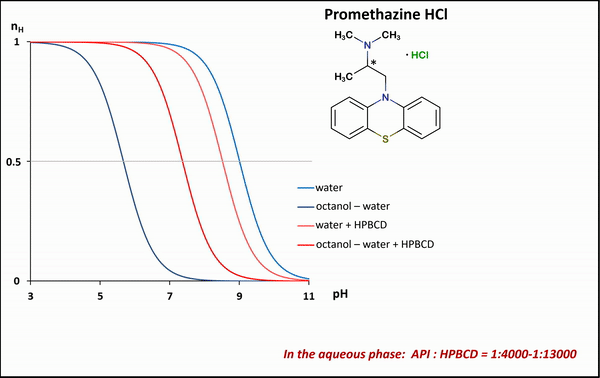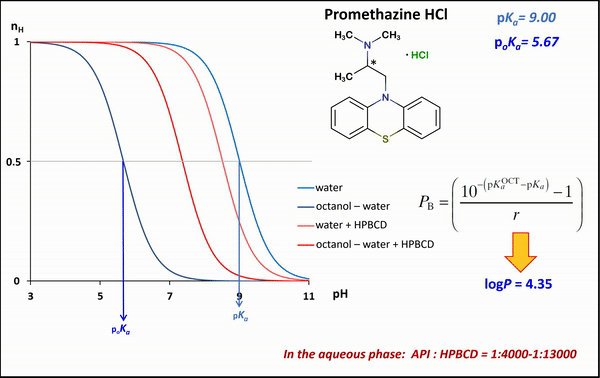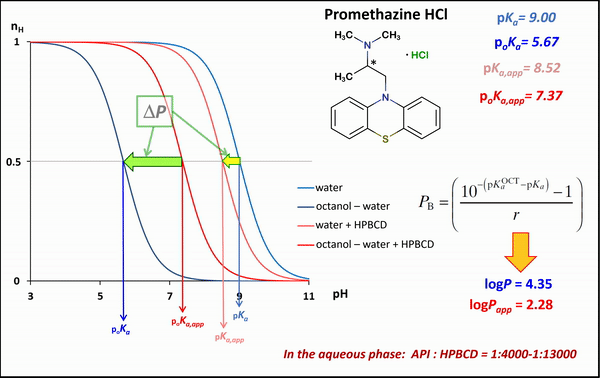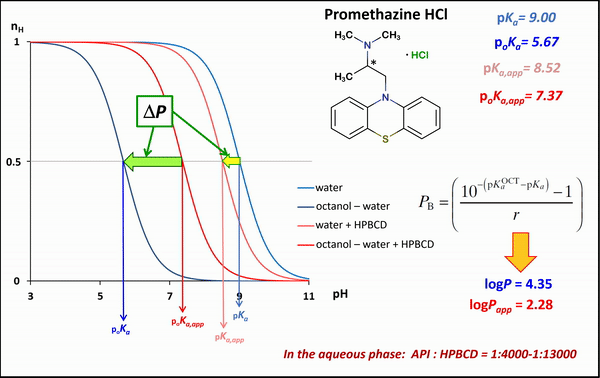
Figure 4 Bjerrum plots of potentiometric differential titration in water, octanol-water system in the presence and absence of HPBCD
After the determination of apparent coefficients at various HPBCD concentrations, their reciprocal could be plotted as a function of HPBCD concentration (Fig. 5). From the slope of the line fitted onto the data points the complex stability constant of the drug–HPBCD complex (K1:1,CD) could be calculated using the following equations:
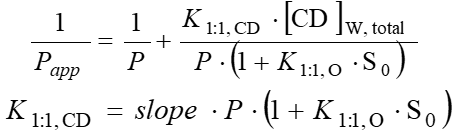
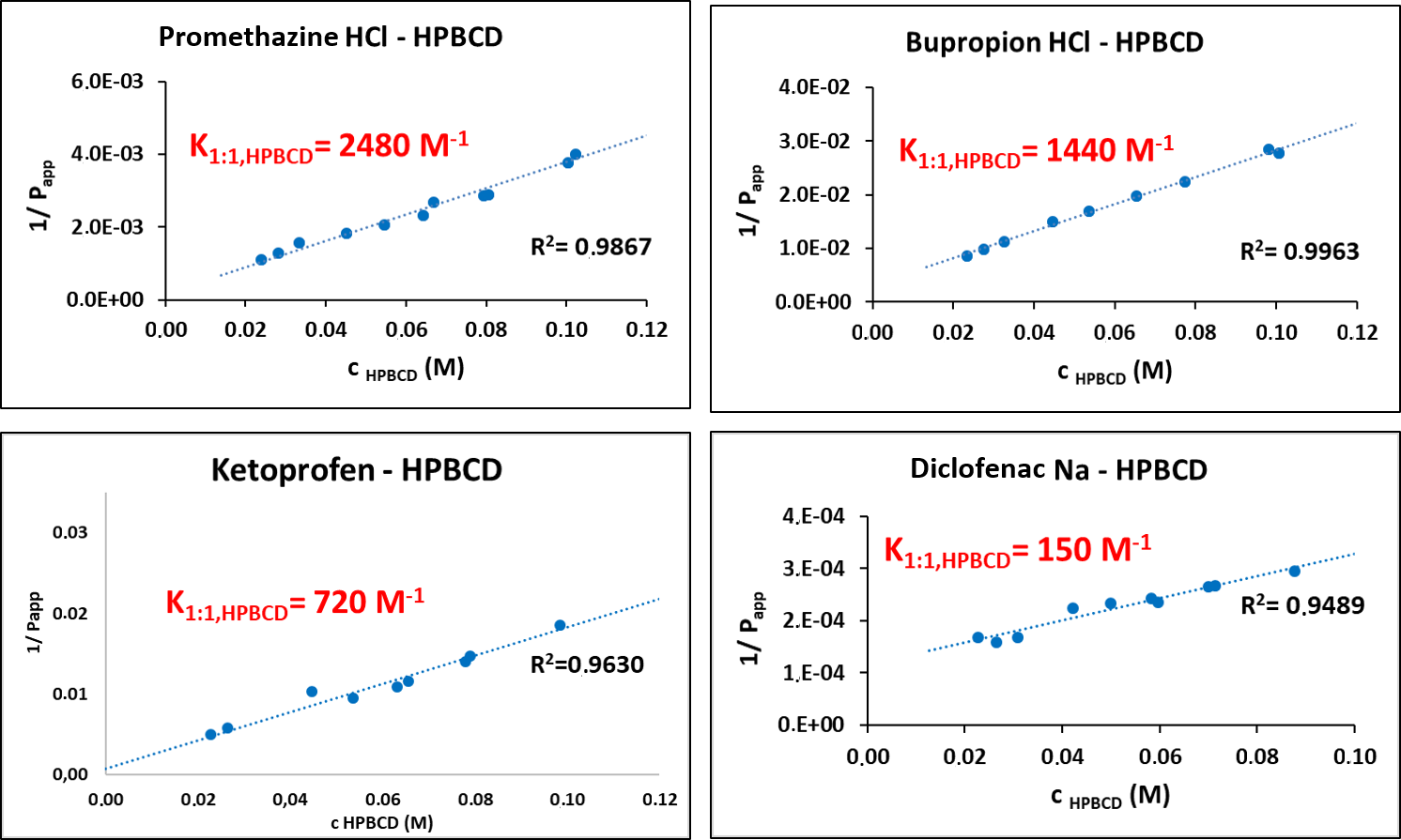
Figure 5 Complex stability constants calculated from changes of partition coefficients of the 4 investigated APIs.
Comparing the K1:1,CD values obtained by our potentiometric method and various other methods it could be observed that they are of the same order of magnitude, therefore we can conclude that our method is applicable for the determination of the stability constants of the drug–CD complexes [P5].
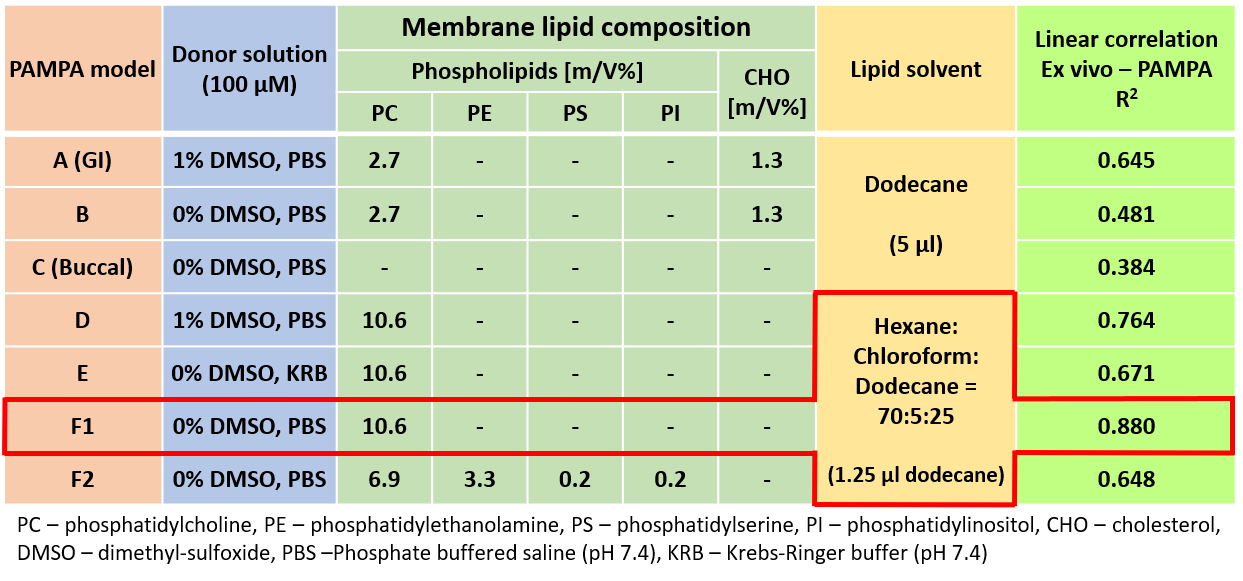
2)
To find the best measurement conditions for the prediction of corneal
permeability, various models were investigated (Table 1). The optimal model
proved to be the one without cosolvent, using phosphatidylcholine dissolved
in a mixture of hexane- dodecane-chloroform as an artificial membrane (Model
F1). Further optimization of the lipid membrane to simulate the lipid
composition of the cornea did not improve the model (Model F2).
Table 1 Results of optimization steps of the corneal-PAMPA model
Using the optimized model for permeability measurements of eye drops we demonstrated its applicability for ophthalmic formulations. The results show that by 5-, 10- and 20-fold dilutions we could simulate the diluting mechanism of tears and blinking (Fig. 6) [P4].
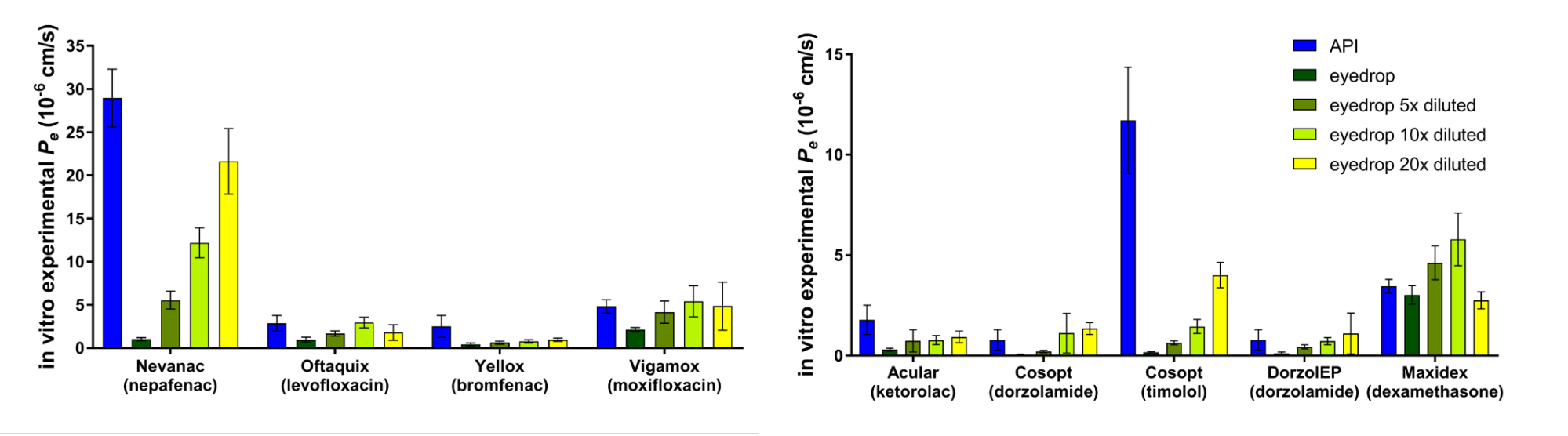
Figure 6 Results of testing the corneal-PAMPA model on commercially
available eye drops.
Testing the corneal-PAMPA model on a large number of drugs, we found that based on solely physicochemical descriptors, the prediction of corneal permeability is not possible as precisely as with our in vitro model [P3]. Based on the experimental permeability data, we also established an in silico prediction model that can be used in the future as a fast and robust way to predict corneal permeability [P6].
3) In the case of APIs with high serum albumin binding affinity, significant changes could be observed in the pKa values measured by UV-pH titration. For APIs with moderate or low binding affinity no significant shift in pKa values could be measured (Table 2). The different pKa values of multiprotic compounds (e.g. nitrazepam) often showed unequal changes that allowed us to assign the place of interaction with HSA. This was also confirmed by the binding modes generated using molecular docking (Figure 7.).
Table 2 Results of the determination of HSA binding affinity by HPLC, rapid equilibrium dialysis and UV-pH titration.
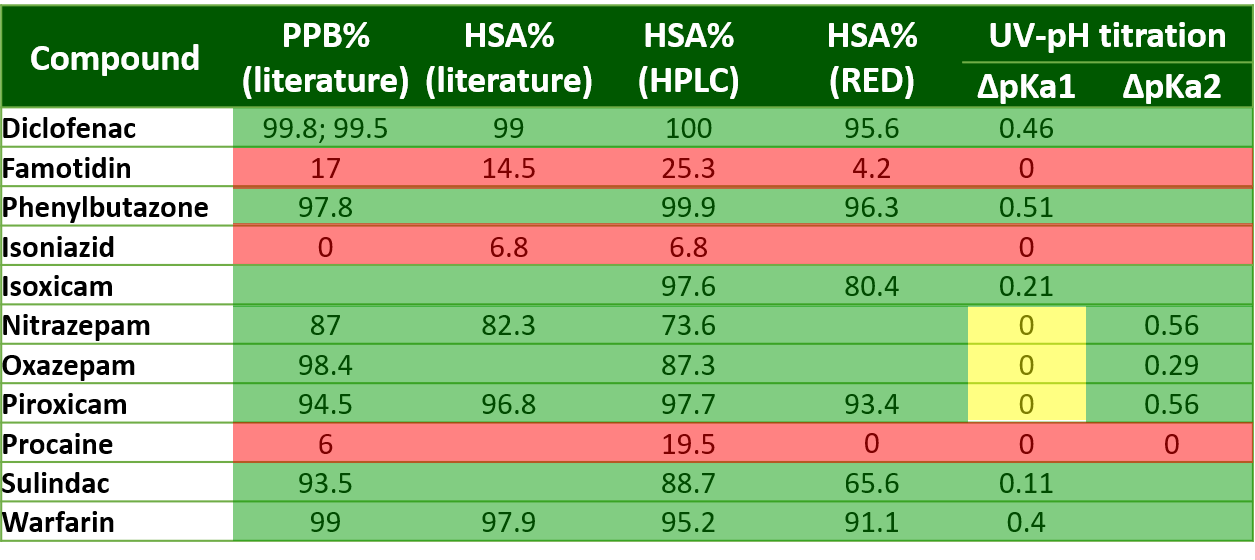
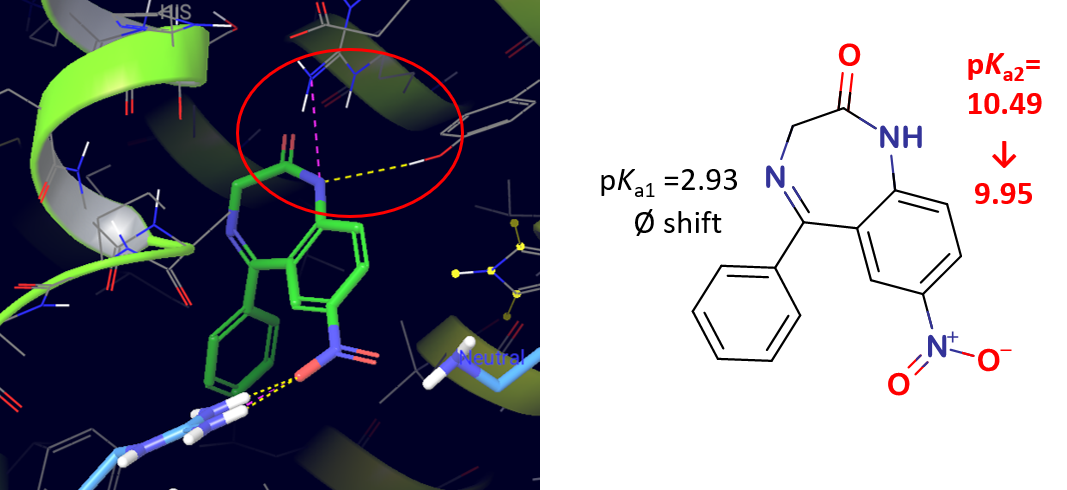
Figure 7 Binding mode generated by molecular docking and pKa shifts during UV-pH titration in the case of nitrazepam
Based on the results, we can conclude that UV-pH titration can be successfully applied for the determination of HSA binding affinity and can provide additional structural information as well.
4) Comparing the dissolution profile and flux of nanoparticles (MEL-HSA-Tween) containing HSA, meloxicam (MEL) and surfactant (Tween 80) in an optimized ratio with the results obtained in the case of nanoparticles without surfactant (MEL-HSA) and MEL powder, we could observe that the nanoparticles have much better pharmacokinetic properties based on both the dissolution profiles and fluxes of the permeability measurements (Fig. 8). The best results were obtained with MEL-HSA-Tween, therefore this formulation is recommended for intranasal administration in the hope of achieving better bioavailability [P2].
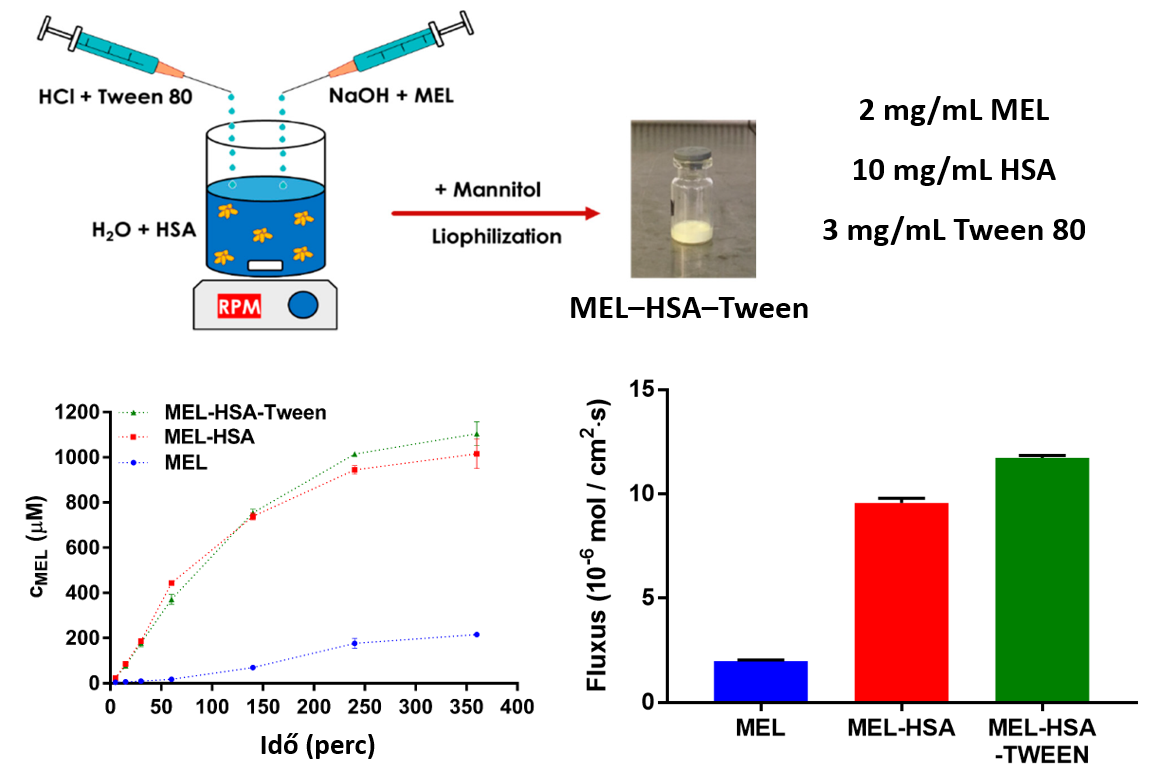
Figure 8 Dissolution profiles and fluxes determined by permeability measurements of HSA nanoparticles (MEL-HSA, MEL-HSA-Tween) and powder meloxicam (MEL)
Expected impact and further research
Our publications and posters on the topics presented here have been received with interest by domestic and international researchers.
Our technique for the determination of complex stability constants using the partition coefficient method can be used as a novel, orthogonal method for CD complexation studies.
Our research group is currently working on the development of therapeutic formulations using the corneal-PAMPA model; we also aim to publish further articles in the topic in the near future [P6, P7].
The UV-pH titration technique may open up the way to support the lead optimization process by filtering out compounds with high HSA binding affinity or by facilitating effective structural modifications to lower HSA binding affinity.
The intranasal administration of meloxicam-containing HSA nanoparticles seems to be a promising alternative for direct, CNS-targeted delivery of drugs, which is also supported by the results of in vivo animal experiments carried out at the University of Szeged.
Publications, references, links
List of corresponding own publications
[P1] Dargó, G., Bajusz, D., Simon, K., Müller, J., Balogh, Gy.T., Human Serum Albumin Binding in a Vial: A Novel UV-pH Titration Method To Assist Drug Design. Journal of Medicinal Chemistry, 2020, 63, 1763–1774. IF = 6.205
[P2] Katona, G., Balogh, Gy.T., Dargó, G., Gáspár, R., Márki, Á., Ducza, E., SztojkovIvanov, A., Tömösi, F., Kecskeméti, G., Janáky, T., Kiss, T., Ambrus, R., Pallagi, E., Szabó-Révész, P., Csóka, I., Development of meloxicam-human serum albumin nanoparticles for nose-to-brain delivery via application of a quality by design approach. Pharmaceutics, 2020, 12, 97. IF = 4.421
[P3] Vincze, A., Dargó, G., Balogh Gy. T., Cornea-PAMPA as an orthogonal in vitro physicochemical model of corneal permeability, Periodica Polytechnica Chemical Engineering, 2020, 64 (3), 384-390. IF = 1.257
[P4] Dargó, G., Vincze, A., Müller, J., Kiss, H. J., Nagy, Z. Zs., Balogh, Gy. T., Corneal-PAMPA: A novel, non-cell-based assay for prediction of corneal drug permeability, European Journal of Pharmaceutical Sciences, 2019, 128, 232-239. IF: 3.616
[P5] Dargó, G., Boros, K., Péter, L., Malanga, M., Sohajda, T., Szente, L., Balogh, Gy. T., Novel medium-throughput technique for investigating drug-cyclodextrin complexation by pH-metric titration using the partition coefficient method, International Journal of Pharmaceutics, 2018, 542 (1-2), 100-107. IF: 3.645
[P6] Vincze, A., Dargó, G., Rácz, A., Balogh, Gy. T., A corneal-PAMPA-based in silico model for predicting corneal permeability, before submission
[P7] Dargó, G., Szilágyi, B. Á., Gyarmati, B., Budai Szűcs, M., Szilágyi, A., Balogh, Gy. T., Synthesis, complex formation and corneal permeation of mucoadhesive cyclodextrin-modified thiol-functionalized poly(aspartic acid) formulations of dexamethasone, in preparation
List of further own publications
[FP1] Szabó-Szentjóbi, H., Márton, A., Pál, D., Dargó, G., Szigetvári, Á., Szántay, Cs., Balogh, Gy. T., Tóth, T., Huszthy, P., Synthesis, Fluorescence and NMR Spectroscopic Studies of a Novel Phosphinoxido-18-crown-6 Ether Containing an Anthracene Fluorophore Unit, Periodica Polytechnica Chemical Engineering, 2019, 64:(1), 37-45. IF = 1.257
[FP2] Nagy, S., Fehér, Zs., Dargó, G., Barabás, J., Garádi, Zs., Mátravölgyi B., Kisszékelyi, P., Dargó, Gy., Huszthy, P., Höltzl, T., Balogh, Gy. T., Comparison of Cinchona Catalysts Containing Ethyl or Vinyl or Ethynyl Group at Their Quinuclidine Ring, Materials, 2019, 12:(18), 3034. IF = 3.057
[FP3] Könczöl, Á., Dargó, G., Brief overview of solubility methods: Recent trends in equilibrium solubility measurement and predictive models, Drug Discovery Today: Technologies, 2018, 27, 3-10.
[FP4] Dargó, G., Bölcskei, A., Grün, A., Béni, Sz., Szántó, Z., Lopata, A., Keglevich, Gy., Balogh, Gy. T., Proton dissociation properties of arylphosphonates: Determination of accurate Hammett equation parameters, Journal of Pharmaceutical and Biomedical Analysis, 2017, 143, 101-109. IF: 3.255
[FP5] Németh, T., Dargó, G., Petró, J. L., Petrik, Zs., Lévai, S., Krámos, B., Béni, Z., Nagy, J., Balogh, Gy. T., Huszthy, P., Tóth, T., Synthesis and pKa determination of new enantiopure dimethyl-substituted acridino-crown ethers containing a carboxyl group: Useful candidates for enantiomeric recognition studies, Chirality: The Pharmacological Biological and Chemical Consequences of Molecular Asymmetry 2017, 29 (9), 522-535. IF: 1.956
[FP6] Borbás, E., Sinkó, B., Tsinman, O., Tsinman, K., Kiserdei, E., Démuth, B., Balogh, A., Bodák, B., Domokos, A., Dargó, G., Balogh, Gy. T., Nagy, Zs. K., Investigation and mathematical description of the real driving force of passive transport of drug molecules from supersaturated solutions, Molecular Pharmaceutics, 2016, 13 (11), 3816-3826. IF: 4.440
[FP7] Kupai, J., Kisszékelyi, P., Rojik, E., Dargó, G., Hegedűs, L., Bezzegh, D., Maszler, P., Szabó, L., Németh, T., Balogh, Gy. T., Huszthy, P., Synthesis and determination of pKa values of new enantiopure pyridino- and piperidino-18-crown-6 ethers” Arkivoc 2016, (IV), 130-151. IF: 1.031
[FP8] Szabó, T., Dargó, G., Szentjóbi, H., Tóth, T., Krámos, B., Izrael, R., Oláh, J., Németh, T., Balogh, Gy. T., Huszthy, P., Synthesis, experimental and theoretical studies on the factors influencing the pKa values of new crown ethers containing a diarylphosphinic acid unit, Tetrahedron, 2016, 72 (52), 8593-8602. IF: 2.651
[FP9] Dargó, G., Balogh, Gy. T., pH-metriás titrálás a fizikai-kémia alapú gyógyszerkémia szolgálatában: ciklodextrin-gyógyszermolekula komplex-képzés vizsgálata (gyors) UV-pH titrálással, Magyar Kémiai Folyóirat - Kémiai Közlemények (1997-) 2016 122 (2-4), 117-123.
[FP10] Müller, J., Esső, K., Dargó, G., Könczöl, Á., Balogh, Gy. T., Tuning the predictive capacity of the PAMPA-BBB model. European Journal of Pharmaceutical Sciences, 2015. (79), 53-60. IF: 3.756
Table of links.
List of references
[1] Szente L. Ciklodextrinek: Nanoméretű konténerektől a terápiás eszközökig, Magyar Kémiai Folyóirat - Előadások, 2015., 121. évf., 1. szám, 34-38.
[2] Natalie L. Trevaskis, Susan A. Charman, Ravi M Shanker, William N. Charman, Colin W. Pouton, Christopher J. H. Porter, Hywel D. Williams, Strategies to Address Low Drug Solubility in Discovery and Development, Pharmacological Reviews, 2013, vol. 65, pp. 315-499.
[3] T. Bohnert and L.-S. Gan, Plasma Protein Binding: From Discovery to Development, Journal of Pharmaceutical Sciences, 2013, vol. 102, no. 9, pp. 2953–2994.
[4] Dujuan Lu, Stephen G. Weber, Zhi Chen, High-Throughput Phase-Distribution Method to Determine Drug-Cyclodextrin Binding Constants, Journal of Pharmaceutical Sciences, 2009, vol. 98, 229-238.
[5] K. A. Connors, Binding Constants: The Measurement of Molecular Complex Stability, Wiley-Interscience, 1987.
[6] M.Kansy, F. Senner, K. Gubernator, Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes, Journal of Medicinal Chemistry, 1998,41(7), 1007-1010.
[7] K. Valkó, S. Nunhuck, C. Bevan, M. H. Abraham, D. P. Reynolds, Fast gradient HPLC method to determine compounds binding to human serum albumin. Relationships with octanol/water and immobilized artificial membrane lipophilicity., Journal of Pharmaceutical Sciences, 2003, 92(11), 2236-2248.
[8] N. J. Waters, R. Jones, G. Williams, B. Sohal, Validation of a Rapid Equilibrium Dialysis Approach for the Measurement of Plasma Protein Binding, Journal of Pharmaceutical Sciences, 2008, 97(10), 4586-4595.