|
|
BMe Kutatói pályázat |
|

Rádai Zita
BMe kutatói pályázat - 2018
III. díj
![]()
Oláh György Doktori Iskola
Szerves Kémia és Technológia Tanszék
Témavezető: Dr. Keglevich György
α-Hidroxifoszfonátok környezetbarát szintézisének kidolgozása és citotoxicitás-vizsgálata
A kutatási téma néhány soros bemutatása
Az α-hidroxifoszfonátok és származékaik jelentősége biológiai hatásukban rejlik. Elsősorban enziminhibitorként1, baktericidként2,3, növényvédőszerként4, illetve antioxidánsként5,6 ismertek. A szerteágazó alkalmazási területüknek köszönhetően a vegyületcsalád újabb, vélhetően bioaktív tagjainak előállítása egy örökzöld kutatási terület. Ahogy a finomkémia és vegyipar minden területén, úgy az α-hidroxifoszfonátok és származékaik előállítása során is egyre inkább előtérbe kerülnek a „zöldkémiai” megfontolások7.

A kutatóhely rövid bemutatása
A Szerves Kémia és Technológia Tanszék, Környezetbarát és Foszforkémiai Kutatócsoportjában elsősorban környezetbarát szintézismódszerek kidolgozásával, a mikrohullámú (MW) technika szerves kémiai alkalmazásával, ipari jelentőséggel bíró reakcióutak újragondolásával és racionalizálásával, katalizátorok tesztelésével és új foszforvegyületek előállításával foglalkozunk Dr. Keglevich György tanszékvezető egyetemi tanár irányítása alatt.
A kutatás történetének, tágabb kontextusának bemutatása
Az α-hidroxifoszfonátok legáltalánosabb előállítási módja oxovegyületek és dialkil-foszfit reakciója. Ennek a reakcióútnak a felfedezése Pudovik nevéhez fűződik, és az 1950-es évekre nyúlik vissza8. Ekkor még katalizátorként drága és erős bázisokat alkalmaztak, illetve a reakciót magas hőmérsékleten, hosszú reakcióidő alatt hajtották végre.
Az utóbbi évtizedekben számos publikáció látott napvilágot, amely az α-hidroxifoszfonátok környezetbarát előállítását célozta9-12. A környezettudatosság jegyében ezek a módszerek arra helyezték a hangsúlyt, hogy a kémiai reakciót oldószermentes körülmények között valósítsák meg. A kémiai reakció során esetlegesen alkalmazott oldószermennyiség azonban sokszor csak a „jéghegy csúcsát” jelenti (1. ábra). Nem hagyható figyelmen kívül az a tény, hogy a termék tisztítását valamilyen formában meg kell oldani. A reakcióelegyben a kiindulási anyagok, a katalizátor, a várt termék és az esetleges melléktermékek egyszerre vannak jelen, és a nekünk szükséges komponens tiszta formában való kinyerése sokszor kihívást jelentő feladat a szintetikus szerves kémiában. A termék izolálására általánosan bevett módszerek az oszlopkromatográfia, az extrakció, valamint az átkristályosítás, amelyek nagy mennyiségű, sokszor illékony és tűzveszélyes szerves oldószerek felhasználását igénylik. Az irodalmi eljárások szerint az α‑hidroxifoszfonátok tisztítását sok esetben ezek kombinációjával oldották meg9-12. Így annak ellenére, hogy a kémiai reakció során nem volt jelen oldószer, a feldolgozási lépések nagy oldószerigénye miatt mégsem beszélhetünk környezetbarát megvalósításokról. Ipari szemmel nézve, az oldószer-felhasználás csökkentése különösen nagy jelentőségű, mind környezetvédelmi, mind gazdasági szempontból.

1. ábra: A szerves kémiában leggyakrabban alkalmazott feldolgozási módszerek oldószerigény szerinti csökkenő sorrendben. Ehhez képest a kémiai reakció oldószerigénye csak a „jéghegy csúcsa”
A kutatás célja, a megválaszolandó kérdések
Kutatásunk első lépése arra irányult, hogy újragondoljuk az α-hidroxifoszfonátok előállítását zöldkémiai szemmel, és a vegyületcsaládot egy olyan módszerrel tegyük hozzáférhetővé, ahol nem csak a reakció, hanem a feldolgozás során is minimális az oldószer-felhasználás.
Célunk volt az α-hidroxifoszfonátok továbbalakítása különféle szerves kémiai reakciókban, melyek szintén bioaktív vegyületekhez vezetnek. Az egyik előállítani kívánt vegyületcsalád az α-aminofoszfonátok voltak (2. ábra, B reakcióút), melyek biológiai hatása a fehérjeépítő α-aminosavakkal való szerkezeti hasonlóságukban rejlik. Terveink között szerepelt ezen kívül α-hidroxifoszfonsavak előállítása (2. ábra, C reakcióút), illetve α‑hidroxifoszfonátok foszforilezési reakcióinak vizsgálata (2. ábra, D reakcióút). Ez utóbbi terület egy teljesen új, eddig nem vizsgált reakcióút. A szintézisutak kidolgozásán kívül a vizsgált kémiai reakciók mechanizmusára is kíváncsiak voltunk.
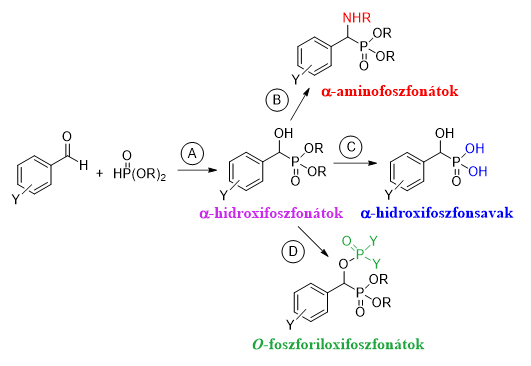
2. ábra: α-Hidroxifoszfonátok és az általunk előállítani kívánt származékok szintézisterve
Annak ellenére, hogy az α-hidroxifoszfonátok és származékaik számos biológiai hatása ismert az irodalomból, citotoxicitásukat még alig, konkrétan egyetlen tanulmányban vizsgálták13. Célul tűztük ki az általunk előállított vegyületek bioaktivitásának tesztelését egy választott rákos sejtvonalon.
Módszerek
Az α-hidroxifoszfonátok előállítását a 2. ábra alapján benzaldehidből és dialkil-foszfitból kiindulva hajtottuk végre (2. ábra, A reakcióút). Katalizátorként egy olcsó, szerves bázist, a trietilamint (Et3N) alkalmaztuk. Új, „zöld” eljárásunk szerint a reakcióelegyet kis mennyiségű acetonban forraltuk, majd a reakcióelegyhez egy kicsapó oldószert, pentánt adtunk. Így hűtés hatására a várt termék kikristályosodott a reakcióelegyből, és egy egyszerű szűréssel megkaptuk az α-hidroxifoszfonátokat (3. ábra)R1,R3. A módszer előnye, hogy a szerves oldószerek oldatban tartják a katalizátort és az esetlegesen visszamaradt kiindulási anyagokat és melléktermékeket, így a kivált α-hidroxifoszfonát-kristályok nagy tisztaságúak. A módszer fő újdonsága, hogy az irodalmi példákkal ellentétben nincs szükség a termék utólagos tisztítására. Bár a kémiai reakció során használtunk kis mennyiségű szerves oldószert, a tisztítási lépések kiiktatásával a teljes felhasznált oldószermennyiséget lényegesen csökkentettük az irodalmi módszerekhez képest.

3. ábra: Az általunk kidolgozott módszer α-hidroxifoszfonátok előállítására
Az α-hidroxifoszfonát-kristályok szerkezetét egykristály-röntgendiffrakciós mérésekkel vizsgáltuk – többek között. Ez egy olyan szerkezetvizsgálati módszer, melynek segítségével pontosabb képet kaphatunk a kristályrács felépítéséről. A módszer azt a jelenséget használja ki, hogy a röntgensugarak szóródnak a molekulák elektronjain, a diffrakciós képek sokaságából pedig akár az atomi koordináták is kiszámíthatók.
Az α-aminofoszfonátokat α-hidroxifoszfonátok és primer aminok reakciójában állítottuk elő (4. ábra). A reakció elősegítéséhez a MW besugárzást hívtuk segítségülR2.

4. ábra: α-Aminofoszfonátok előállítása MW reaktorban.
A laboratóriumi reaktor a konyhai MW sütők elvén működik. A hagyományos víz- vagy olajfürdős melegítéssel ellentétben a mikrohullámok közvetlenül a molekulákkal lépnek kölcsönhatásba, ezáltal nem kell számolni a reakcióedény falán át történő hőátadás lassú folyamatával. A MW besugárzás hatására a dipólusmomentummal rendelkező (poláris) molekulák oszcillálni kezdenek. A molekulák súrlódása lokális túlmelegedési pontok („hot spotok”) kialakulását eredményezi (5. ábra), melyek segítik átlendíteni a reakciót akár egy magasabb entalpiagáton. Így MW körülmények között sokszor olyan reakciók is lejátszódnak, melyek hagyományos melegítés hatására nem mennek végbe14.
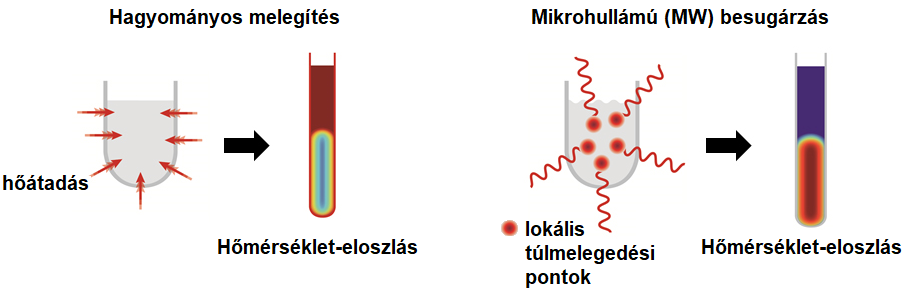
5. ábra: Hagyományos és MW fűtés elméleti háttere és hőmérséklet-eloszlása
Az α-hidroxifoszfonsavak előállítását a megfelelő dibenzil-észterek katalitikus hidrogénezésével valósítottuk meg egy nyomásmérővel ellátott autoklávban (6. ábra).
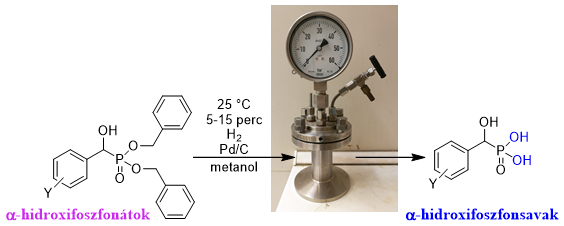
6. ábra: α-Hidroxifoszfonsavak előállítása katalitikus hidrogénezéssel
Az O-foszforiloxifoszfonátokat α-hidroxifoszfonátok és savkloridok reakciójában állítottuk elő (7. ábra).
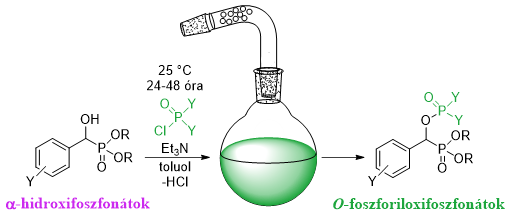
7. ábra: O-foszforiloxifoszfonátok előállítása
A reakciók mechanizmusát több esetben számításos kémiai módszerekkel tanulmányoztukR2. A kvantumkémiai számítások célja, hogy a kiindulási anyagok, a termék és az ezeket összekötő átmeneti állapot energiáját és térszerkezetét meghatározza. A kiindulási anyagok és a termék lokális energiaminimumban elhelyezkedő, a valóságban is izolálható vegyületek, ezzel szemben az átmeneti állapot csak egyetlen molekularezgés idejéig (nagyságrendileg 10-13 s) létezik, amelynek vizsgálatára nem létezik rutinszerűen alkalmazható kísérleti módszer, ellenben kvantumkémiai módszerekkel jól modellezhető (8. ábra). Ezek az elméleti módszerek abban különböznek egymástól, hogy egy adott, atommagok és elektronok sokaságából álló rendszer energiájának számításához milyen elhanyagolással élnek. A számításokat Gaussian03 programcsomaggal, DFT (density functional theory – sűrűségfunkcionál-elmélet) alkalmazásával, B3LYP/6-31G(d,p), illetve B3LYP/6-31++G(d,p) elméleti szinteken végeztük.
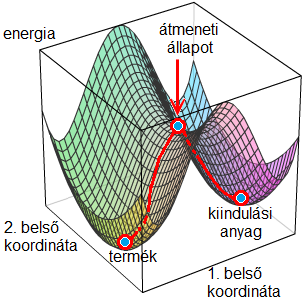
8. ábra: Potenciálisenergia-felület. A piros görbe jelöli a kémiai reakció útvonalát (belső koordináták lehetnek például kötéshosszak, kötésszögek)
A citotoxicitás-vizsgálatok automatizált, robotkaros készülékben (Hamilton StarLet) történtek. A vegyületek citotoxicitását 200 μM koncentrációban teszteltük olyan humán szarkóma rákos sejteken (Mes-Sa), melyek genetikai módosítások következtében egy fluoreszcens fehérjét (mCherry) termelnek. A fluoreszcencia – mely fluoriméterrel mérhető – egyenes arányban áll az élő sejtek számával, melyből a hatóanyag által elpusztított sejtek aránya számolható.
Eddigi eredmények
A kutatómunka során egy új módszert dolgoztunk ki α-hidroxifoszfonátok előállítására oxovegyületek és dialkil-foszfit reakciójában, trietilamin katalizátor jelenlétébenR1,R3. A módszer fő újdonsága, hogy az oldószer-felhasználást a minimumra szorítottuk, így az irodalmi módszereknél kevesebb illékony szerves oldószerrel terheljük a környezetet. A módszer további előnye, hogy kiváló termeléssel (78–95%) juthatunk az α‑hidroxifoszfonátokhoz. A reakcióval egybekötött kristályosítási lépésnek köszönhetően a várt termékeket nagy tisztaságban (> 99%) kaptuk.
A kísérleti tapasztalatok azt mutatták, hogy a reakció körülményei között, trietilamin katalizátor nélkül nem keletkezett a várt α-hidroxifoszfonát. Annak érdekében, hogy alátámasszuk a trietilamin reakcióban betöltött katalitikus szerepét, kvantumkémiai számításokkal vizsgáltuk a reakció mechanizmusát. A számítások szerint trietilamin nélkül a reakció aktiválási entalpiagátja 85,9 kJ/mol, míg a katalizátor jelenlétében ez az érték 68,8 kJ/mol-ra csökken. A számítások rámutattak arra, hogy a trietilamin úgy vesz részt az aktiválási entalpiagát csökkentésében, hogy segíti a proton-átadást a foszfitról az oxovegyület karbonilcsoportjára, így lehetővé teszi a kémiai reakció lejátszódását (9. ábra)R2.
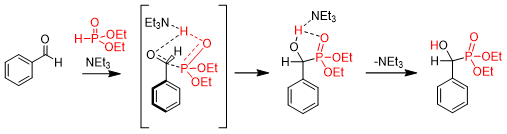
9. ábra: Trietilamin-katalizált Pudovik-reakció mechanizmusa kvantumkémiai számítások alapján
Az egykristály-röntgendiffrakciós mérések szerint a kristályszerkezetben kétféle elrendeződési módja lehet az α-hidroxifoszfonát molekuláknak. Bizonyos esetekben a rácspontokban dimerek találhatók, míg más származékoknál a molekulák láncokat képeznek. Mindkét esetben a képződmények fő összetartó ereje a H-híd. Az általunk vizsgált vegyületek esetében azt a tendenciát tapasztaltuk, hogy az α-helyzetű CH3‑csoport jelenléte a dimerek keletkezésének kedvez, míg az α-metilcsoport hiányában a láncszerű képződmények kialakulása a kedvezményezett (10. ábra).
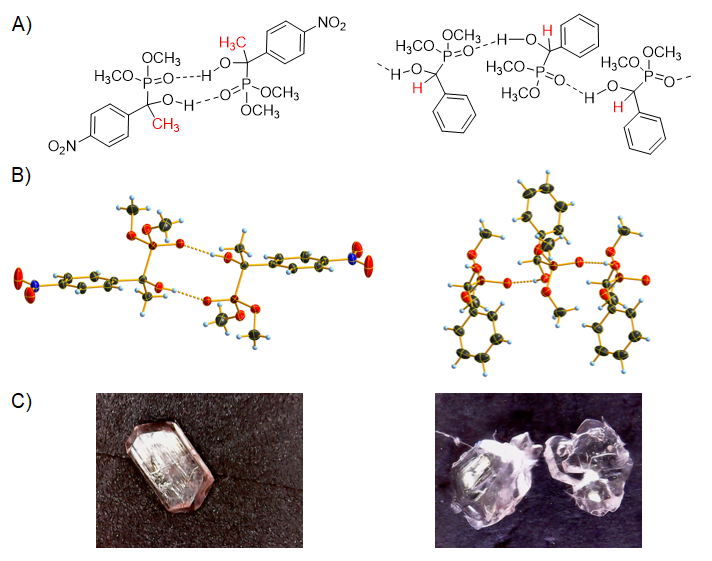
10. ábra: α-Hidroxifoszfonátok szerkezetvizsgálata egykristály-röntgendiffrakciós mérésekkel. A) α-Hidroxifoszfonát dimerek és láncasszociátumok szemléltetése. B) Röntgenfelvételek az α-hidroxifoszfonát-kristályokról. C) Fénymikroszkóppal készült fényképek az α-hidroxifoszfonát-egykristályokról
Az α-aminofoszfonátok előállítása során meglepődve tapasztaltuk, hogy a sztérikusan gátolt α-helyzetű hidroxicsoport szubsztitúciója igen könnyen, 15–30 perc alatt lejátszódottR2. Ez arra sarkalt minket, hogy a mechanizmusnak ismét kvantumkémiai számításokkal járjunk utána. A szokatlanul nagy reakciókészségnek az az oka, hogy a nukleofil szubsztitúciót egy kedvező szomszédcsoport-hatás segíti. Vagyis a nagy energiájú P–O‑kötés létesülésével egyidejűleg a Cα–O kötés meggyengül, ami megkönnyíti az amin nukleofil támadását (11. ábra)R2.
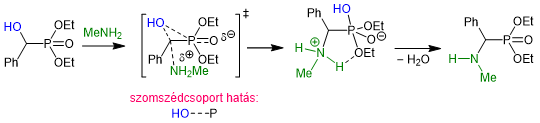
11. ábra: Az α-aminofoszfonátok keletkezését egy kedvező szomszédcsoport hatás segíti
A citotoxicitás-vizsgálatok során α-hidroxifoszfonsavakat, α-hidroxifoszfonátokat és O-foszforiloxifoszfonátokat vizsgáltunk (12. ábra). A szerkezeti képletek alatt található számok százalékban kifejezve azt mutatják, hogy 48 óra inkubálás után a kontroll (hatóanyagot nem tartalmazó) kísérlethez képest mennyivel kevesebb élő sejtet tartalmazott az a minta, amely 200 μM koncentrációban tartalmazta az adott hatóanyagot. A nullaközeli érték azt jelenti, hogy a hatóanyag nem gátolta a sejtek osztódását, azaz nem volt hatásos a vegyület. A száz százalékos érték pedig a rákos sejtek teljes pusztulását jelentette. A 12. ábrán látható, hogy az aromás gyűrűket tartalmazó származékok voltak igazán hatásosak a Mes-Sa sejtek ellen.
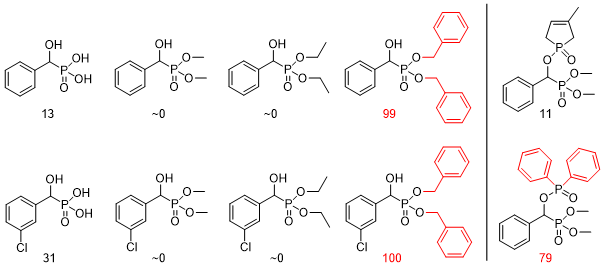
12. ábra: Szerkezet-hatás összefüggések az α-hidroxifoszfonsavak, α-hidroxifoszfonátok és O-foszforiloxifoszfonátok citotoxicitás-vizsgálata során
Várható impakt, további kutatás
A kutatómunka során ipari és környezetvédelmi szemszögből is körüljártuk az α‑hidroxifoszfonátok előállítását. A felesleges oldószer-felhasználás kiiktatásával és a feldolgozás egyszerűsítésével egy olyan módszert dolgoztunk ki, amely jó kiindulópontja lehet akár egy ipari szintézisnek is. Az α-hidroxifoszfonátokat különféle szerves kémiai reakciókban hasznosítottuk, és ezzel új vegyületcsaládokat tettünk hozzáférhetővé.
Az α-hidroxifoszfonátok és származékaik számos biológiai hatása ismert, azonban rákos sejtvonalakon korábban alig vizsgálták őket. Az általunk előállított vegyületkönyvtár citotoxicitás-vizsgálata egy úttörő kutatás. Az eddigi mérések biztató eredményekkel kecsegtetnek, azonban a szerkezet-hatás összefüggések mélyebb megértéséhez a későbbiekben még tervezzük új vegyületek előállítását és tesztelését. A rákellenes szerek terápiás alkalmazhatóságának egyik kulcskérdése, hogy a vegyületek az egészséges sejtekre milyen hatással vannak, aminek felderítése szintén további terveink részét képezi.
Saját publikációk, hivatkozások
A pályázat témájához közvetlenül kapcsolódó saját publikációk
Folyóiratcikkek:
R1. Keglevich, G.; Rádai, Z.; Kiss, N. Z. To date the greenest method for the preparation of α-hydroxyphosphonates from substituted benzaldehydes and dialkyl phosphites. Green Process. Synth. 2017, 6, 197-201. [IF: 0,736]
R2. Kiss, N. Z.; Rádai, Z.; Mucsi, Z.; Keglevich G. Synthesis of α-aminophosphonates from α-hydroxyphosphonates; A theoretical study. Heteroatom Chem. 2016, 27, 260-268. [IF: 1,221]
Rövid közlések:
R3. Rádai, Z.; Kiss, N. Z.; Mucsi, Z.; Keglevich, G. Synthesis of α-hydroxyphosphonates and α-aminophosphonates. Phosphorus, Sulfur Silicon Relat. Elem. 2016, 191, 1564–1565. [IF: 0,809]
R4. Rádai, Z. Zöld módszerekkel a biológiailag aktív anyagokért: Egy értékes vegyületcsalád előállítása. Élet és tudomány, 2018, 10, 303–305. [IF: -]
Könyvfejezet:
R5. Rádai, Z.; Kiss, N. Z.; Keglevich, G. Synthesis of α-hydroxyphosphonates, an important class of bioactive compounds. In: Keglevich György (szerk.), Organophosphorus Chemistry: Novel developments. 315 p. Berlin; Boston: Walter de Gruyter, 2018, 91–107. ISBN: 978-3-11-053453-5 [IF: -]
R6. Rádai, Z.; Kiss N. Z.; Keglevich, G. Chalcogenides: Advances in Research and Application (NOVA Science Publishers, New York, USA), 2018, közlésre elfogadva. [IF: -]
Összefoglaló (review) cikk:
R7. Rádai, Z.; Keglevich, G. Synthesis and reactions of α-hydroxyphosphonates. Molecules, 2018, 23, 1439. [IF: 3,098 (2017)]
A pályázat témájához közvetlenül nem kapcsolódó saját publikációk
Folyóiratcikkek:
R8. Keglevich, G.; Rádai, Z.; Harsági, N.; Szigetvári, Á.; Kiss, N. Z. A study on the acidic hydrolysis of cyclic phosphinates: 1-Alkoxy-3-phospholene 1-oxides, 1-ethoxy-3-methylphospholane 1-oxide, and 1-ethoxy-3-methyl-1,2,3,4,5,6-hexahydro-phosphinine 1-oxide. Heteroatom Chem. 2017, 28, e21394. [IF: 1,137]
R9. Kiss, N. Z.; Rádai, Z.; Tihanyi, I.; Szabó, T.; Keglevich, G. Microwave-assisted direct esterification of a cyclic phosphinic acid with phenols. Mendeleev Commun. 2018, 28, 31–32. [IF: 2,098 (2017)]
R10. Kiss, N. Z.; Rádai, Z.; Mucsi, Z.; Keglevich, G. The synthesis of bis(phosphinoyl)amines and phosphinoyl–phosphorylamines by the N-phosphinoylation and N-phosphorylation of 1-alkylamino-2,5-dihydro-1H-phosphole 1-oxides. Heteroatom Chem. 2015, 26, 134–141. [IF: 1,203]
Rövid közlések:
R11. Kiss, N. Z.; Rádai, Z.; Keglevich, G. Derivatization of phosphinic acids in the presence of ionic liquids. Phosphorus, Sulfur Silicon Relat. Elem. 2016, 191, 1494–1496. [IF: 0,809]
R12. Kiss, N. Z.; Mucsi, Z.; Rádai, Z.; Böttger, É. V.; Keglevich, G. The synthesis and potential use of cyclic phosphinic acid derivatives. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 668–671. [IF: 0,723]
Összefoglaló (review) cikk:
R13. Rádai, Z.; Kiss, N. Z.; Keglevich G. An overview of the applications of ionic liquids as catalysts and additives in organic chemical reactions. Curr. Org. Chem. 2018, 22, 533–556. [IF: 2,193 (2017)]
Hivatkozások listája
1. Patel, D. V.; Rielly-Gauvin, K.; Ryono, D. E.; Free, C. A.; Rogers, W. L.; Smith, S. A.; DeForrest, J. M.; Oehl, Jr. R. S.; Petrillo, E. W. J. Med. Chem. 1995, 38, 4557-4569.
2. Pokalwar, R. U.; Hangarge, R. V.; Maske, P. V.; Shingare, M. S. Arkivoc, 2006, 11, 196–204.
3. Kategaonkar, A. H.; Pokalwar, R. U.; Sonar, S. S.; Gawali, V. U.; Shingate, B. B.; Shingare, M. S. Eur. J. Med. Chem., 2010, 45, 1128–1132.
4. Song, H.; Mao, H.; Shi, D. Chin. J. Chem., 2010, 28, 2020-2024.
5. Rao, K. U. M.; Sundar, C. S.; Prasad, S. S.; Rani, C. R.; Reddy, C. S. Bull. Korean Chem. Soc. 2011, 32, 3343–3347.
6. Naidu, K. R. M.; Kumar, K. S.; Arulselvan, P.; Reddy, C. B.; Lasekan, O. Arch. Pharm. Chem. Life Sci. 2012, 345, 957–963.
7. Olszewski, T. K. Synthesis, 2014, 46, 403-429.
8. Pudovik, A. N.; Zametaeva, G. A. Izvestiya Akademii Nauk. S.S.S.R., Seriya Khimicheskaya, 1952, 932–939.
9. Nandre, K. P.; Nandre, J. P.; Patil, V. S.; Bhosale, S. V. Chem. Biol. Interface, 2012, 2, 314–321.
10. Kumar, K. S.; Reddy, C. B.; Reddy, M. V. N.; Rani, C. R.; Reddy, C. S. Org. Commun., 2012, 5, 50–57.
11. Aouani, I.; Lahbib, K.; Touil, S. Medicinal Chem., 2015, 11, 206-213.
12. Ramananarivo, H. R.; Solhy, A.; Sebti, J.; Smahi, A.; Zahouily, M.; Clark, J.; Sebti, S. ACS Sustain. Chem. Eng., 2013, 1, 403–409.
13. Kalla, R. M. N.; Lee, H. R.; Cao, J.; Yoo, J. W.; Kim, I. New J. Chem., 2015, 39, 3916–3922.
14. Keglevich, G.; Kiss, N. Zs.; Mucsi, Z.; Körtvélyesi, T. Org. Biomol. Chem. 2012, 10, 2011–2018.