|
|
BMe Kutatói pályázat |
|

Kollár Levente
BMe kutatói pályázat - 2022
![]()
Oláh György Doktori Iskola
BME Vegyészmérnöki és Biomérnöki Kar, Szerves Kémia és Technológia Tanszék
Témavezető: Dr. Keserű György Miklós
Kovalens inhibitorok fejlesztése változatos gyógyszercélpontokra
A kutatási téma néhány soros bemutatása
Egyes kovalens inhibitorként működő hatóanyagok már hosszú évtizedek óta gyógyszerkincsünk részét képezik; e hatásmechanizmust követő molekulák kutatása mégis csak az ezredforduló tájékán kezdett egyre nagyobb teret nyerni. Ennek oka, hogy alkalmazásuk akár kockázatokat is hordozhat, ám a gyógyszerkutatók az elmúlt évtizedben elkezdték felismerni és értékelni a bennük rejlő felbecsülhetetlen terápiás lehetőségeket. A teljesség igénye nélkül, a kovalens inhibitorok előnyei közé sorolhatjuk általánosságban nagy hatékonyságukat, hosszú tartózkodási idejüket, jó biokémiai hasznosulásukat, valamint alkalmazásukkal olyan célfehérjék is megcélozhatóvá válnak, melyek tisztán nemkovalens kölcsönhatások kialakítása révén nem támadhatók. Munkánk során több terápiás szempontból fontos szerepet játszó fehérjével szemben is célul tűztük ki kovalens inhibitorok azonosítását és fejlesztését.
A kutatóhely rövid bemutatása
Jelen kutatás az Eötvös Loránd Kutatási Hálózat (ELKH) részét képző Természettudományi Kutatóközpontban (TTK), a Gyógyszerkémiai Kutatócsoportban zajlik, melynek vezetője Dr. Keserű György Miklós akadémikus, egyetemi tanár. A csoport kiterjedt hazai, valamint nemzetközi kapcsolathálóval rendelkezik, a kialakult együttműködések keretében nagyszámú gyógyszerkutatási programban vagyunk érdekeltek. Kovalens inhibitorok kutatásával 2015 óta foglalkozunk.
A kutatás történetének, tágabb kontextusának bemutatása
Kovalens inhibitornak nevezzük azokat a vegyületeket, melyek hatásukat a célpontként szolgáló fehérjék meghatározott nukleofil aminosav-oldalláncaival kialakított kovalens kötés kialakítása révén hozzák létre. A gyógyszerkutatás történetét szemlélve kijelenthetjük, hogy hosszú időn keresztül távolságtartás övezte ezeket a vegyületeket. Ez bizonyos szempontból érthető, ugyanis az említett nagy reaktivitású kovalens inhibitorok alkalmazása esetén emelkedett immunogenitási és toxicitási kockázattal számolhatunk a nemkovalens hatásmechanizmusú hatóanyagokhoz képest [1,2].
A képet ugyanakkor árnyalja, hogy például az egyik legrégebb óta biztonsággal alkalmazott gyógyszerünk, az aszpirin is kovalens inhibitorként fejti ki hatását, valamint a β-laktám típusú antibakteriális szerek – melyek első képviselője a penicillin – szintén ezen hatásmechanizmust követik (1. ábra). A paradigmaváltás a 2000-es évek elejére tehető, amikor a gyógyszerkutatók egyre inkább felismerték, és elkezdték helyén kezelni a kovalens inhibitorokban rejlő lehetőségeket. A kovalens gátlás folyamata kétlépéses mechanizmust követ: az első lépés a molekuláris felismerés, mely során a ligandum nemkovalens kölcsönhatások kialakításával stabilizálódik a kötőhelyen, ezt követi a kovalens kötés kialakítása az adott aminosav-oldallánccal. Ebből következik nagy előnyük, a kiemelkedő hatékonyság: teljes gátlás is elérhető, ráadásul a terápiás hatás jellemzően alacsonyabb dózis alkalmazásával kiváltható. A nemegyensúlyi kötődés eredményeképp alkalmazásuk a farmakokinetikai jellemzőkre kevésbé érzékeny. Külön kiemelendő továbbá, hogy a kovalens megközelítés olyan célfehérjék esetén is fegyvert jelent, melyeknél a kötőhely felszíni, avagy nagyon flexibilis, ezek csupán nemkovalens kölcsönhatások kialakításával nem támadhatók [3].
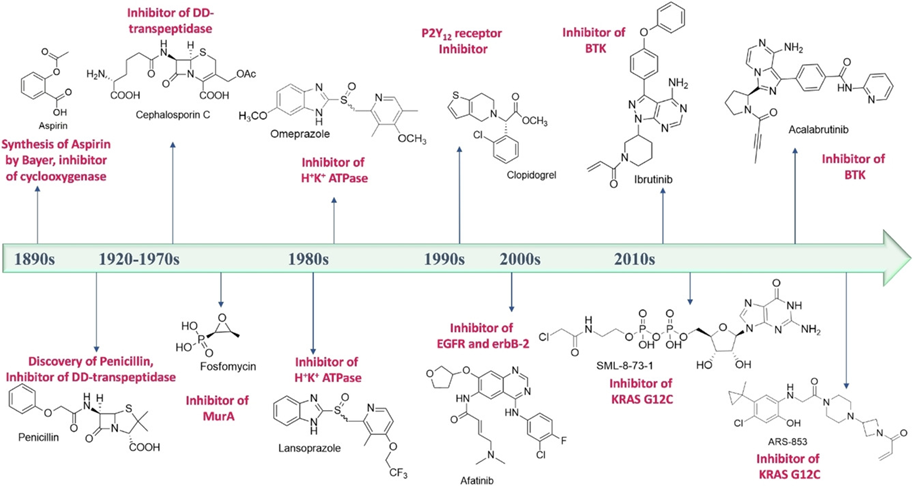
1. ábra: A jelenleg is használt legfontosabb kovalens inhibitorok, az idővonalon követhető a felfedezésük sorrendje [4]
A kutatás célja, a megválaszolandó kérdések
Munkánk során célul tűztük ki kovalens inhibitorok fejlesztését különböző fehérjecélpontokra, amivel bizonyítható a megközelítés sokoldalúsága.
Kiemelt célpontunknak az immunoproteaszómát (iPS) választottuk, mely multikatalitikus fehérjekomplex, feladata a sérült, valamint rosszul feltekeredett fehérjék lebontása; hibás működését több onkológiai, autoimmun, valamint neurodegeneratív betegséggel is összefüggésbe hozták. A különböző immunoproteaszóma alegységek különféle katalitikus aktivitással rendelkeznek, szelektív gátlásuk feltétlenül kívánatos a potenciális mellékhatások visszaszorítása érdekében. Az alegységek strukturális hasonlósága miatt a szelektív inhibitorok azonosítása kihívásokkal teli, azonban a β5i-alegység a kötőhelyén fellelhető specifikus cisztein aminosavnak köszönhetően kovalens célzásra ad lehetőséget. Munkánk során β5i-szelektív immunoproteaszóma inhibitorok azonosítását és ezek fejlesztését kívántuk megvalósítani [5,6].
A penicillinkötő fehérjék (PBP) olyan enzimek, melyek alapvetően a baktériumok peptidoglikán bioszintézisében vesznek részt, ezáltal kulcsszerepet töltenek be a bakteriális sejtfal felépítésének folyamatában. A penicillinkötő fehérjék gátlása jellemzően β-laktám típusú vegyületekkel (pl.: penicillin) valósítható meg, ugyanakkor napjainkban egyre nagyobb problémát jelent a baktériumok rezisztenciájának kialakulása, amelynek kezeléséhez további baktériumellenes szerek kutatása és fejlesztése kívánatos. Célul tűztük olyan új antibakteriális szerek azonosítását, melyekben kovalens kötés kialakításáért felelős reaktív csoport (warhead) nem a hagyományos β-laktám, hanem egyéb szerin-reaktív kötőelem [7,8].
A COVID19-járvány az egész világ működését alapjaiban borította fel. A pandémia leküzdése az egész emberiség közös érdekévé és feladatává lépett elő. Hamar kiderült, hogy a koronavírus főproteázának (3CLPro) kulcsszerepe van a vírus replikációjában, gátlása előnyös stratégiának bizonyult, így az egyik elsőszámú gyógyszercélponttá lépett elő. Az említett fehérje a cisztein-proteázok közé tartozik, ennek megfelelően alkalmas kovalens inhibitorokkal történő célzásra [9,10].
Módszerek
Az említett gyógyszerkutatási projekteket hazai és nemzetközi partnereinkkel közösen végezzük. Csoportunk felelős a munka számításos kémiai háttérrel történő támogatásáért, valamint a számítógépes gyógyszertervezés eredményeként kapott molekulák szintéziséért, ezek és fehérjekomplexeik analitikai és biofizikai jellemzéséért. Mindhárom célfehérje esetén a Ljubljanai Egyetem kutatóival dolgozunk együtt, akik az elkészült vegyületek biokémiai aktivitását vizsgálják. A kovalens inhibitorok kutatásában elengedhetetlenül fontos a vegyületek fehérjével való kötődésének bizonyítása. Ebben kulcsszerepe van a tömegspektrometriai méréseknek, melyben a TTK MS Metabolomika Kutatólaboratórium van segítségünkre. Amennyiben az adott molekula igazoltan jelölni képes a fehérjét, a kapott konjugátum megfelelő enzimekkel történő emésztése révén információt nyerhetünk arról, hogy a kismolekula pontosan melyik aminosav oldalláncával alakított ki kötést.
Az immunoproteaszóma inhibitorok kutatása során a már említett szelektivitási kihívás mellett további nehézséget jelent, hogy az irodalomban ismert inhibitorok nagy hányada peptid vagy peptidomimetikum szerkezetű, melyek gyakran gyenge metabolikus stabilitást vagy alacsony orális biohasznosulást eredményezhetnek. Ezt kiküszöbölendő, fragmens-szűrést végeztünk, mely során több száz fragmensméretű molekula (kisméretű, egyszerű szerkezetű, poláris molekula) aktivitását mértük egy célszerűen választott koncentrációban az immunoproteaszóma β5i-alegységével szemben, majd a legaktívabb vegyületek esetén IC50-értéket is meghatároztunk.
Az aktívnak bizonyult szerkezetek környezetében vegyülettárakat építettünk, ezeket vizsgáltuk szerkezet–hatás összefüggések keretében, így jutottunk három analóg vegyületcsaládhoz: benzoxazol-2-tion, benzimidazol-2-tion és benzotiazol-2-tionhoz (együttesen: benzXazol-2-tionok), ezeket tovább optimáltuk. Halogén-léptetést végeztünk, így vizsgáltuk a fragmensek kémiai módosításának előnyös irányait, mely alapján több tucat vegyület vásárlására és szintézisére került sor. A vásárolt vegyület megfelelő tisztaságát HPLC és 1H NMR segítségével ellenőriztük, és amennyiben szükséges volt, tovább tisztítottuk őket. A szintetizált vegyületek az irodalomban szinte kivétel nélkül eddig ismeretlenek voltak, így minden esetben teljes karakterizálást (HPLC, 1H NMR, 13C NMR, HRMS, olvadáspont) végeztünk. Az inhibíció mechanizmusának feltérképezésére biofizikai módszereket és biokémiai teszteket használtunk.
Az új antibakteriális szerek (PBP1b-inhibitorok) kutatása keretében több szerin-kötő szerkezeti elemet tartalmazó könyvtárat is biokémiai assay-nak vetettünk alá: teszteltünk karbamátokat, boronsavakat és boronésztereket, valamint különféle N-metilezett heterociklusokat. Akadályt jelentett, hogy a cisztein-kötésre képest vegyületek esetén a biológiai teszt komponenseivel interferencia lépett fel, így egy új, megfelelő protokoll is kidolgozásra került. Számításokat végeztünk kovalens inhibitorok kötődési mechanizmusának feltárására és az affinitást meghatározó szerkezeti elemek és tulajdonságok azonosítására. Több olyan boronsavat szintetizáltunk, melyek modellezés alapján képesek lehetnek több kötőzseb egyszerre történő kitöltésére, ezáltal erős gátlás kialakítására.
A nagy nemzetközi érdeklődésnek köszönhetően a 3CLPro kismolekulákkal való kölcsönhatásával kapcsolatban rövid idő alatt nagy mennyiségű adat (pl. Röntgen-krisztallográfiás szerkezet) keletkezett, amely felhasználható új 3CLPro-inhibitorok racionális tervezésére. Munkánk során új módszertani megoldások (pl. fragmens-összekötés kísérletes kötőmódok alapján) segítségével igyekszünk hatékony, új 3CLPro-inhibitorokat tervezni és szintetizálni. Emellett a korábban, egyéb fehérjecélpontokon tesztelt vegyületeink szűrésével (pl. benz-X-azol könyvtár) is sikerült új 3CLPro-gátlószereket azonosítanunk, amelyek hatásmechanizmusát magas szintű molekulamodellező módszerekkel szeretnénk értelmezni.
Eddigi eredmények
Fragmensszűrés (2. ábra) eredményeképp azonosítottunk három rokon vegyületcsaládot, amelyek az immunoproteaszóma β5i-alegységén kötődő, nagy ligandumhatékonyságú inhibitoroknak bizonyultak.
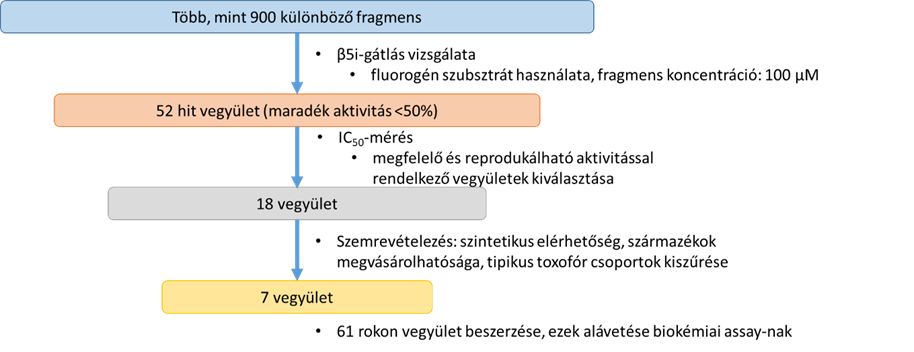
2. ábra: Fragmensszűrés folyamata
A fragmensalapú gyógyszerkutatás stratégiáját követve ezeket a benz-X-azol-vázas vegyületcsaládokat elsőként az alapvázak klórral egyszeresen szubsztituált származékaiként vizsgáltuk (klór-szken). Tekintettel arra, hogy a leghatékonyabb gátlást a 6-os, valamint 7-es pozícióban szubsztituált analógok mutatták, a továbbiakban ilyen származékot állítottunk elő és vetettük alá biokémiai tesztelésnek (3. ábra). Összehasonlítottuk továbbá a benzimidazol-2-tion-vegyületek és N-metilezett analógjaik aktivitását. Utóbbiak jellemzően jóval hatásosabbak voltak, így számos N-metil-benzimidazol-2-tion származékot szintetizáltunk és teszteltünk. Előállítottunk továbbá egy újabb szériát, melyben a 6-klórbenzimidazol-2-tion alapváz a N-atomon jellemzően kisméretű, poláris szubsztituenseket hordoz. Összesen 68 származék szintézisére került sor [S1].
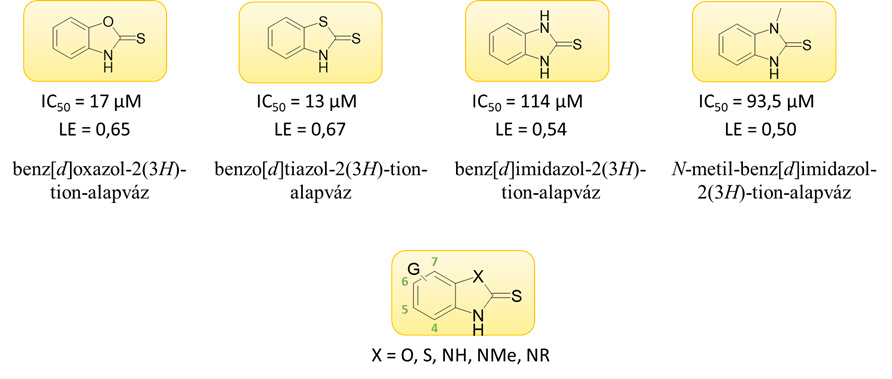
3. ábra: Az azonosított alapvázak szerkezete a biológiai aktivitás és a ligandumhatékonyság társaságában (felül); a szintetizált származékok általános képlete, az alapváz számozása jelölve (alul)
A kovalens kötőelem optimalizálását is elvégeztük: az azonosított vegyületekben egységesen jelenlévő tion-csoportot cseréltük más cisztein-reaktív szerkezeti egységekre. Ennek eredményeként azonosítottuk a karbonitril-származékokat, melyek aktivitási, szelektivitási és reaktivitási szempontok összességét tekintve előnyösebbnek kínálkoztak a tionoknál. Benzoxazol-2-karbonitril alapvázú vegyületekből 27 tagú szisztematikusan szubsztituált könyvtárat építettünk (4. ábra), teszteltük a vegyületek stabilitását, valamint modellvegyületekkel (glutation, N-acetil cisztein) szembeni reaktivitásukat. Meghatároztuk a biológiai aktivitásukat az immunoproteaszóma β5i-alegységével szemben; a leghatásosabb vegyületek esetén szelektivitást is vizsgáltunk. Csoporthatékonyság-analízist végeztünk, az alapváz legelőnyösebb szubsztituált pozícióinak vizsgálata céljából.
Vizsgálódásaink során összefüggést kerestünk a vegyületek reaktivitása és biokémiai aktivitása közt, némi meglepetésre azonban nem találtunk a kettő közt kirajzolódó trendet. Ez a tény is rávilágít arra, hogy fragmensméretű kovalens inhibitorok esetén is kulcskérdés a nemkovalens felismerés [S2].
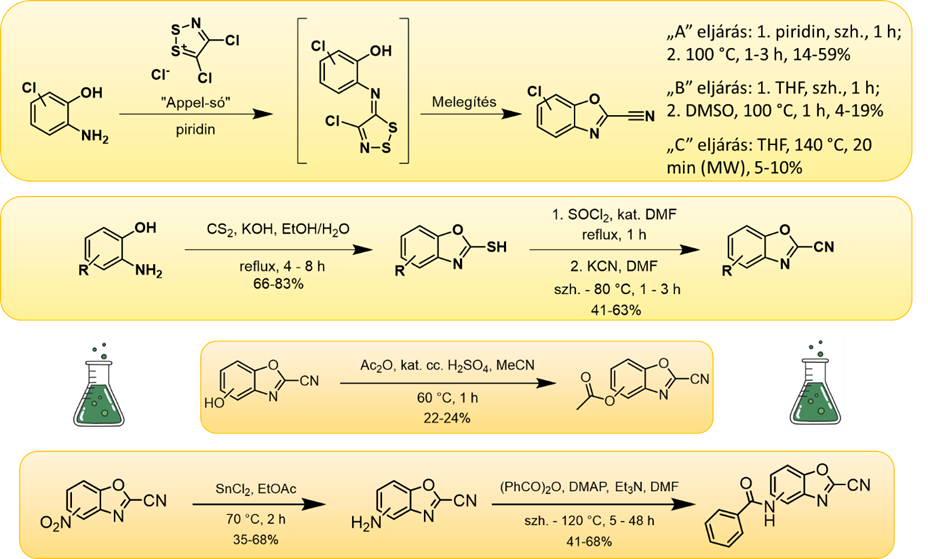
4. ábra: A benzoxazol-2-karbonitril vegyülettár felépítéséhez általánosan használt reakciók
A legígéretesebb vegyületek 2–4 µM közötti IC50 értékkel (β5i) rendelkeztek. Foglalkoztunk olyan molekulák szintézisével is, melyek egy távtartón (linkeren) keresztül összekötve tartalmazzák a Bortezomibban is fellelhető (R)-boroleucin molekularészletet, valamint a benzoxazol-2-karbonitril vagy a benzimidazol-2-karbonitril alapvázat: ennek eredményeként lehetőség szerint „kétfogú” (két kovalens kötés kialakítására is képes) vegyületként képesek működni (5. ábra). 12 származék szintézisére került sor (6. ábra), a leghatásosabb molekula esetén sikerült szubmikromoláris IC50 értéket (β5i-alegység) elérni [S2].
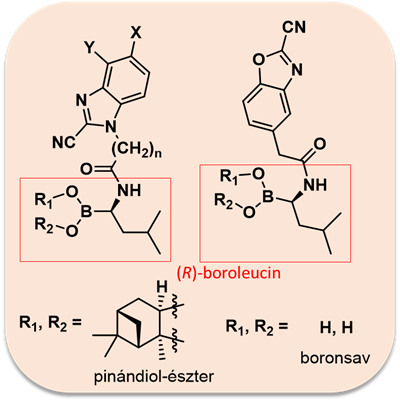
5. ábra: A „kétfogú” vegyületek általános szerkezete
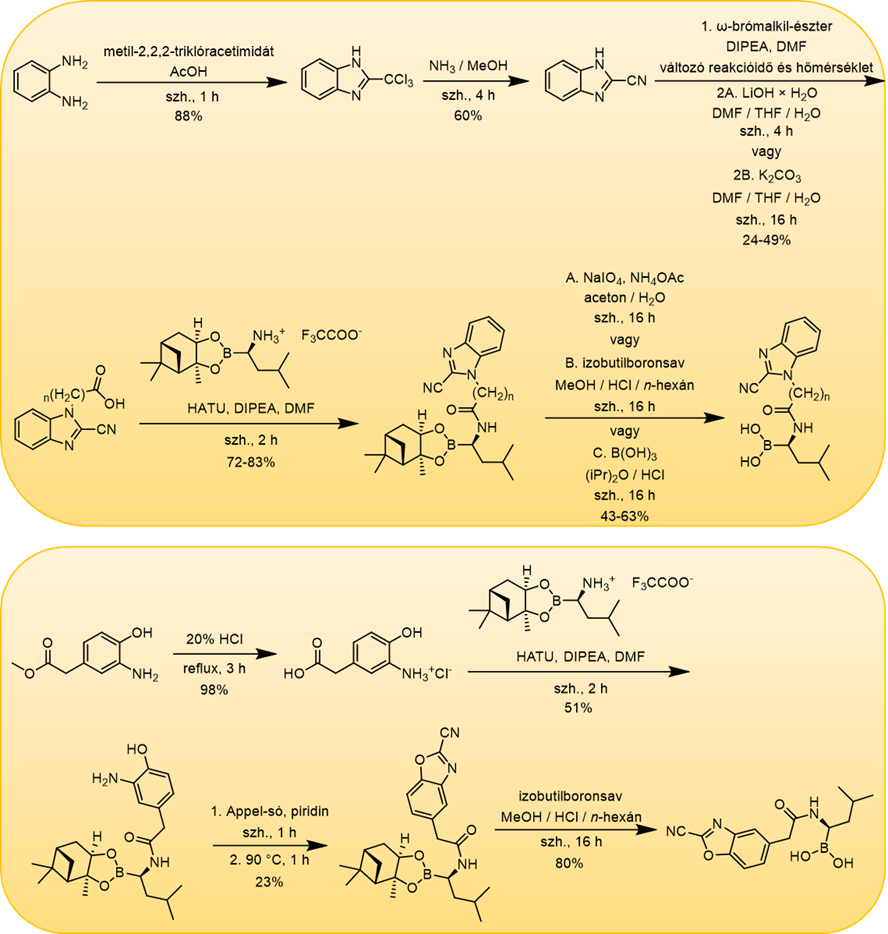
6. ábra: A „kétfogú” vegyületek szintézise
Az immunoproteaszóma β5i-alegységén kötődő inhibitorok azonosítását más megközelítésből is megkíséreltük. 404 kereskedelmi forgalomban kapható, 5-, valamint 6-tagú ciklikus boronsavat vetettünk alá virtuális szűrésnek. A legjobb eredményeket elérő vegyületek közül 12-t biokémiai assay-ban vizsgáltunk: ezek mérhető aktivitással rendelkeztek, az irodalomban elsőként leírt ciklikus boronsav immunoproteaszóma-inhibitornak számítanak [S3].
A PBP1b-inhibitorok kutatásának keretében együttműködő partnereink kidolgoztak egy új biokémiai assay-t, mellyel megvalósítható a cisztein-reaktív vegyületek vizsgálata is. Sikeresen szintetizáltunk és azonosítottunk továbbá két olyan boronsavat, melyek számottevő aktivitással rendelkeznek (IC50 értékek: 77 µM és 50 µM). Ezek a vegyületek kiindulópontul szolgálhatnak további optimalizálási kísérleteknek.
A koronavírus főproteázzal (3CLPro) szemben teszteltük a korábban előállított benz-X-azol könyvtárat, több inhibitort is azonosítottunk. A kapott eredmények alapján további származékok szintézisére került sor, számításos kémiai módszerekkel behatóbban tanulmányoztuk gátlószerek hatásmechanizmusát.
Várható impakt, további kutatás
Az immunoproteaszóma inhibitorok kutatása során szerteágazóan vizsgált benzoxazol-2-karbonitrilek további optimalizálása az eddigi eredmények ismeretében célszerűnek kínálkozik. A tervezett fragmensnövesztési kampányt alapozza meg az elvégzett csoporthatás-analízis. Az előállított kétfogú vegyületek legaktívabb képviselői ugyan nem tekinthetők szelektívnek (pán-proteaszóma-inhibitorok), rákterápiás hatóanyagként történő fejlesztésük ennek ellenére racionális lehet.
A PBP1b-gátlószerek kutatásában eddig elért eredményeket a közeljövőben tervezzük publikálni, továbbá még szélesebb körű szűrést fogunk folytatni a lehetséges reaktív csoportok körében. További lehetőséget kínál a boronsavak optimálása.
A 3CLPro fehérjére azonosított benz-X-azol alapvázú inhibitorok hatásmechanizmusát vizsgáló közlemény előkészítés alatt áll, hamarosan kiadónak történő benyújtásra kész állapotba kerül. Kutatásunk tárgyát képezi jelenleg is fragmensek összekötése révén új 3CLPro-inhibitorok tervezése és szintézise – ezt tervezzük folytatni.
Saját publikációk, hivatkozások, linkgyűjtemény
Kapcsolódó saját publikációk listája
[S1] Kollár, L., Gobec, M., Szilágyi, B., Proj, M., Knez, D., Ábrányi-Balogh, P., Petri, L., Imre, T., Bajusz, D., Ferenczy, G. G., Gobec, S., Keserű, G. M., Sosič, I. „Discovery of selective fragment-sized immunoproteasome inhibitors”, Eur. J. Med. Chem., 2021, 219, 113455. (IF: 6,514)
[S2] Kollár, L.; Gobec, M.; Proj, M.; Smrdel, L.; Knez, D.; Imre, T.; Gömöry, Á.; Petri, L,; Ábrányi-Balogh, P.; Csányi, D.; Ferenczy, G. G.; Gobec, S.; Sosič, I.; Keserű, G. M. „Fragment-Sized and Bidentate (Immuno)Proteasome Inhibitors Derived from Cysteine and Threonine Targeting Warheads”, Cells, 2021, 10, 3431. (IF: 6,600)
[S3] Kollár, L., Ferenczy, G. G., Proj, M., Gobec, M., Gobec, S., Sosič, I., Keserű, G. M. „Virtual Screening and Biochemical Testing of Borocycles as Immunoproteasome Inhibitors”, Per. Pol. Chem. Eng., 2021, 65(3), 292–298. (IF: 1,257)
Linkgyűjtemény
TTK Gyógyszerkémiai Kutatócsoport
koronavírus főproteáz (3CLPro)
Hivatkozások listája
[1] Singh, J., Petter, R. C., Baillie, T. A., Whitty, A. Nat. Rev. Drug Discov. 2011, 10, 307–317.
[2] Bauer, R. A., Drug Discov. Today 2015, 20, 1061–1073.
[3] Johnson, D.S., Weerapana, E., Cravatt, B.F. Future Med. Chem. 2010, 2, 949–964.
[4] Ghosh, A. K., Samanta, I., Mondal, A., Liu, R. ChemMedChem, 2019, 14, 889–906.
[5] Murata, S., Yashiroda, H., Tanaka, K. Nat. Rev. Mol. Cell Biol., 2009, 10, 104–115.
[6] McCarthy, M. K., Weinberg, J. B. Front. Microbiol., 2015, 6, 21.
[7] Inglis, S. R., Strieker, M., Rydzik, A. M., Dessen, A., Schofield, C. J., Anal. Biochem. 2012, 420, 41–47.
[8] Zervosen, A., Sauvage, E., Frère, J. M., Charlier, P. Luxen, A. Molecules 2012, 17, 12478–12505.
[9] Hoffman, R. L., Kania, R. S., Brothers, M. A., Davies J. F., Ferre, R. A., Gajiwala, K. S., He, M., Hogan, R. J., Kozminski, K., Li, L. Y., Lockner, J. W., Lou, J., Marra, M. T., Mitchell, L. J., Murray, B. W., Nieman, J. A., Noell, S., Planken, S. P., Rowe, T., Ryan, K., Smith, G. J., Solowiej, J. E., Steppan, C. M. Taggart B. J. Med. Chem. 2020, 63(21), 12725–12747.
[10] Mody, V., Ho, J., Wills, Mawri, A., Lawson, L., Ebert, M. C. C. J. C., Fortin, G. M., Rayalam S. Commun. Biol. 2021, 4, 93.