|
|
BMe Kutatói pályázat |
|

Oláh György Doktori Iskola
Richter Gedeon Nyrt.
Témavezető: Dr. Keserű György Miklós
Számításos módszerek a fragmens alapú gyógyszerkutatás támogatására
A kutatási téma néhány soros bemutatása
A gyógyszerkutatás elsődleges célja a betegségek gyógyítása és megelőzése. Egy gyógyszer kifejlesztése azonban hosszú és költséges folyamat, melyet magas kiesési arány jellemez [1]. A fragmensalapú gyógyszerkutatás az utóbbi időben megjelent és elterjedt technológia, mely lehetővé teszi a vegyületek gyors optimálását a gyógyszerkutatásban fontos fizikai-kémiai paraméterek hatékony felügyelete mellett, és a legújabb eredmények alapján képesnek mutatkozik a kiesési arány csökkentésére, valamint korábban nehezen célba vehető fehérjék vizsgálatára [2]. Számításos módszerek a fragmensalapú gyógyszerkutatás minden fázisában eredményesen alkalmazhatók a gyógyszerkémiai munka segítésére és eredményességének növelésére [3]. Munkám során célom fragmens találatok azonosítására alkalmas virtuális szűrési módszerek értékelése és fejlesztése, valamint összekapcsolásra alkalmas fragmensek számítógépes azonosítására és összekapcsolás általi vegyülettervezésre alkalmas módszerek fejlesztése volt, különösen a gyógyszeripar számára kiemelt jelentőséggel bíró G-fehérje kapcsolt receptor (GPCR) fehérjecsaláddal szemben.
A kutatóhely rövid bemutatása
A Richter Gedeon Nyrt. magyarországi központú, specializált gyógyszercég. Generikus termékek gyártása mellett Közép-Európa egyik legnagyobb originális kutatóközpontjával rendelkezik, ahol nőgyógyászati és központi idegrendszeri betegségekre irányuló kutatást végez. Legújabb gyógyszere, a törzskönyvezés alatt álló Cariprazine skizofrénia és mánia indikációkban sikeres fázis III vizsgálatokkal rendelkezik. Munkámat a gyógyszergyár Felfedező Kémiai Kutatólaboratóriumának számításos kémiai csoportjában végeztem.
A kutatás történetének, tágabb kontextusának bemutatása
A fragmensalapú gyógyszerkutatás magában foglalja a molekuláris fragmensek kiválasztási, szűrési és optimálási módszereit. Ezek poláris, kis molekulatömegű és alacsony komplexitású vegyületek [4], melyek optimális kölcsönhatások kialakítására képesek a célfehérjével, így jobb kiindulási pontként szolgálnak, mint a nagy áteresztőképességű szűrésben azonosított gyógyszerszerű találatok (1. ábra). További előnyt jelent a gyógyszerszerű molekulákhoz képest a kémiai tér nagyobb arányú reprezentálása. A fragmensek sajátossága azonban, hogy kisebb affinitással kötődnek a fehérjékhez, így szűrésük általában speciális biofizikai módszerekkel történik, és optimálásuk újszerű megközelítést igényel.
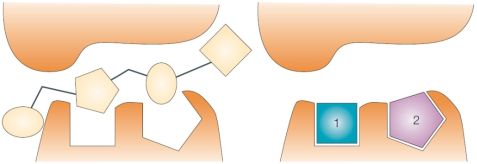
1. ábra. Rossz minőségű HTS találat (bal) és jó minőségű fragmens találatok (jobb) [2].
Mivel a kísérleti szűrési módszerek áteresztőképessége korlátozott, hatékony virtuális fragmens szűrési technikák is szükségesek a biológiailag aktív vegyületek dúsítására. A molekuláris dokkolás a leggyakrabban alkalmazott fehérjeszerkezet-alapú gyors virtuális szűrési módszer, mely segítségével prediktálhatók a kísérleti fragmens kötőmódok [5]. Az irodalomban több sikeres szűrési kampányt is leírtak, az utóbbi időben G-fehérje kapcsolt receptorokkal szemben is [6,7].
Ebbe a családba a hét transzmembrán hélixből álló fehérjék tartoznak, melyek változatos extracelluláris ligandumokat képesek felismerni az ionoktól a kis molekulákon és lipideken keresztül a peptid hormonokig. Szerkezet-meghatározásuk csak az utóbbi években vált elérhetővé, ami egyúttal lehetővé tette a szerkezetalapú gyógyszertervezési módszerek alkalmazását erre a gyógyszerkutatás számára igen fontos fehérjecsaládra is.
A szerkezetalapú fragmens optimálás hatékonyságát mutatja, hogy a Vemurafenib, az első fragmens alapon kifejlesztett gyógyszer (2. ábra) 2011-ben került piacra, és kifejlesztése mindössze 6 évbe telt [8].
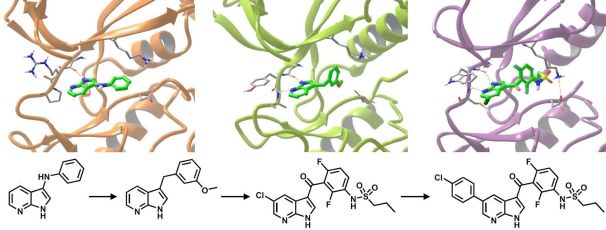
2. ábra. A Vemurafenib optimálása egy azaindol fragmens találatból „növesztés” által [8].
A kutatás célja, a megválaszolandó kérdések
Munkám első részében célom a GPCR célpontokkal szemben történő virtuális fragmens szűrési módszerek értékelése volt. Mivel a GPCR családból egyelőre kevés célpont szerkezete ismert, mind kristályszerkezetek, mind homológia modellek (az aminosav szekvencia és a harmadlagos szerkezet hasonlósága alapján modellezett fehérjeszerkezetek) alkalmazhatóságát vizsgáltam virtuális fragmens szűrésben. A fehérje konformációs flexibilitásának figyelembe vételének hatását is elemeztem mindkét kiindulási információtípus esetén, és összehasonlítottam az egy szerkezetet felhasználó virtuális szűrési módszerekkel. Az eredményeket prospektív fragmensszűrésben használtam fel, és az azonosított fragmenstalálatok egy későbbi optimálási projekt kiinduló pontjai lehetnek.
Munkám második részében összekapcsolásra alkalmas fragmens találatok számítógépes azonosítására alkalmas módszereket vizsgáltam, mivel két fragmens összekapcsolásával kevés lépésben növelhető a kiindulási találatvegyületek affinitása [9]. Ezért az elsődleges (ún. „forró pont”) kötőhelyre történő dokkoláson felül vizsgáltam egy egyszerű szekvenciális dokkolási protokoll hatékonyságát fehérjék másodlagos kötőhelyein kötött fragmensek modellezésében is. A kifejlesztett módszert alkalmaztam a D3 dopamin receptor elsődleges és másodlagos kötőhelyére illeszkedő, összekapcsolásra alkalmas fragmensek azonosítására, valamint szerkezeti alapon magyaráztam az összekapcsolt szintetizált vegyületek szelektivitását a D2 dopamin receptorral szemben.
Módszerek
A gyógyszerkutatásban alkalmazott számításos módszerekkel támasztott egyik legnagyobb követelmény a gyorsaság, hiszen sokszor vegyületek százaira vagy százezreire kell predikciót végezni, amiért cserébe a pontosságuk természetesen limitált. Ezért központi fogalom a dúsulás, mely azt mutatja, mennyivel hatékonyabb csak a megfelelőnek prediktált vegyületek vizsgálata, mint ugyanannyi véletlenszerűen kiválasztott vegyület vizsgálata.
A fehérjék és fehérje-ligandum komplexek méretükből adódóan csak klasszikus mechanikai vagy empirikus számítási módszerekkel modellezhetők. A homológia modellezés során egy fehérje szerkezetét egy másik, ismert szerkezetű, hasonló szekvenciájú fehérje szerkezete alapján modellezhetjük empirikus szabályok és molekulamechanikai konformációkeresés és optimálás segítségével. Fehérjék mozgékonyságának atomi szintű leírására alkalmazhatjuk a szintén erőtéralapú molekuladinamikai (MD) szimuláció módszerét (3. ábra). Ennek során a rendszer időbeli változását követjük a rendszerben ébredő erők számításával és a klasszikus mechanika mozgásegyenletének sokszori megoldásával.
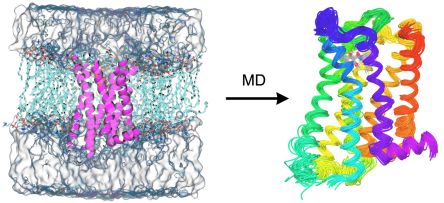
3. ábra. Membránba ágyazott és szolvatált D3 dopamin receptor szerkezete (bal) és a molekuladinamikai szimuláció során kapott szerkezeti sokaság (jobb).
A fehérje-ligandum, jelen esetben fragmens, kölcsönhatást molekuláris dokkolással modellezhetjük, melynek célja a kötő konformáció és a kötődési szabadentalpia becslése. A fragmensek erőtéralapú konformációkeresését dokkolás során a fehérje kötőhelye által meghatározott térben végezzük. A dokkolást végezhetjük merevnek tekintett kötőhelyen, konformációs sokaságon (amely eredhet például molekuladinamikai szimulációból), vagy alkalmazhatunk indukált illeszkedésen alapuló eljárást (IFD), mely figyelembe veszi a fehérje és a ligandum együttes konformációs flexibilitását. A dokkolásalapú virtuális szűrés eredményeit retrospektív esetben a dúsulási faktorokkal, prospektív esetben a találati aránnyal jellemezhetjük.
A fragmens találatok jellemzésére és összehasonlítására a ligandumhatékonysági metrikákat használhatjuk, például az irodalomban elfogadott LE (egy nehéz atom hozzájárulása a kötődési szabadentalpiához), LLE (az affinitás és víz-oktanol megoszlási hányados, a logP közötti elválás mértéke) és LELP (a logP és az LE hányadosa) mérőszámokat. Ez utóbbi lipofil hatékonysági metrikák használatával a vegyületek farmakokinetikai jellemzőit és biztonságosságát jelezhetjük előre [10].
Munkám során a Schrödinger programcsomag Prime modulját használtam ismeretlen szerkezetű GPCR-ek homológia modellezésére, valamint a Glide gyorsdokkoló modult a fragmens ligandumok dokkolására, az ehhez kapcsolódó Python API programozási környezetet a szekvenciális dokkolás automatizálására, a NAMD szoftvert egy együttműködés keretén belül molekuladinamikai szimulációkra, valamint a ChemAxon keminformatikai eszközeit a molekuláris paraméterek számítására és a vegyületkönyvtárak kezelésére.
Eddigi eredmények
Elsődleges kötőhely virtuális szűrés
Elkészültek a H4 hisztamin és az 5HT6 szerotonin receptorok homológia modelljei, továbbá a D3 dopamin és CXCR4 kemokin receptorok röntgen szerkezeteit használtuk fel molekuladinamikai szimulációk kiindulásaként, és a kiindulási szerkezeteken, valamint a kapott konformációs sokaságon dokkolásalapú retrospektív dúsulási teszteket végeztem, melyeket a dúsulási faktorokkal (EF) és a hatásfokmérő karakterisztika görbék alatti területtel (AUC) jellemeztem. Ahogy a 4. ábrán látható, a kiindulási szerkezetek közül a H4 homológia modell volt az egyetlen, amely képes volt elválasztani az aktív és a csali molekulákat, azonban az MD legjobb szerkezetei minden esetben felülmúlták a kiindulási modelleket, függetlenül a célponttól és az alkalmazott kiértékelési módszertől [T1].
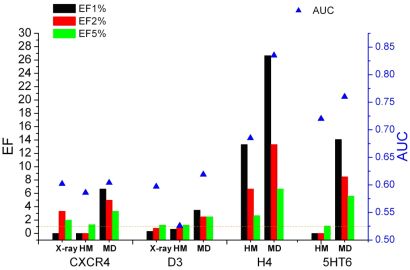
4. ábra. Dúsulási faktorok (színes oszlopok) és AUC értékek (kék háromszögek) a CXCR4, D3, H4, és 5HT6 röntgen szerkezetekre (X-ray), homológia modellekre (HM) és a legjobb molekuladinamikai szerkezetekre (MD).
Ezután a D3 dopamin és H4 hisztamin receptorok szerkezeteivel szemben prospektív virtuális szűrést végeztem a Richter Gedeon fragmenskönyvtárral. Az egyedi kiindulási szerkezetek esetén és szerkezeti sokaságba való dokkolás esetén is kb. 50-50 fragmenst választottam ki biológiai szűrésre. A különböző módszerekkel 16-32%-os valós találati arányt értem el, és azonosítottam kedvező LE (nagyobb mint 0.3) és LELP (kisebb mint 10) értékű, újszerű fragmens találatokat (5. ábra). A röntgenszerkezet és a homológia modell is alkalmasnak bizonyult virtuális szűrésre. A sokaságalapú dokkolás nem mutatott nagyobb hatékonyságot, azonban különböző találatokat adott, így a módszerek kiegészítették egymást [T2].
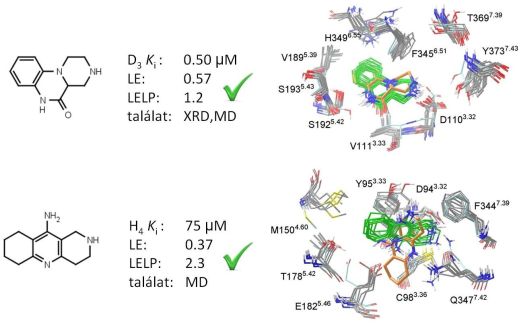
5. ábra. Újszerű fragmens találatok a prospektív fragmensszűrésből, kötődési és ligandhatékonysági adataik valamint prediktált kötőmódjuk az egyedi szerkezetekbe (narancs) és a szerkezeti sokaságba (zöld) történő dokkolás esetén.
Másodlagos kötőhely virtuális szűrés
A munka következő részében egy szekvenciális dokkolási protokoll hatékonyságát elemeztem két vagy több kooperatívan kötött ligandum modellezése végett először 129 kismolekulás fehérje komplex, majd 32 fragmensekkel alkotott fehérje komplex esetében. A kismolekulás adatokból kiderült, hogy két ligandum dokkolása az esetek 55%-ában sikeres, több ligandum dokkolása viszont általában sikertelen. Ha az adatkészletből csak a zárt kötőhelyen kötött gyógyszerszerű molekulákat vizsgáltam, akkor a leghatékonyabb protokollal ez az arány 68%-ra emelkedett. Az adatkészletben szereplő öt, fragmens ligandumokkal kristályosított HSP90 fehérje szerkezetre pedig minden dokkolás sikeres volt, ez ösztönözte a módszer vizsgálatát az új fragmens adatkészleten [T3].
Amint a 6. ábrán látható, a dokkolás sikeres volt minden ligandumra: előálltak 2.0 Å alatti atomi RMSD-vel rendelkező kötőmódok. Ezen túlmenően az esetek 77%-ában 1.0 Å alatti atomi RMSD-vel rendelkezett a dokkolt kötőmód. A kiemelkedő pontosság különösen fontos a virtuális fragmens-összekapcsolás lehetősége szempontjából. Hasonlóan jó eredményeket kaptam keresztdokkolás esetén is, vagyis amikor egy fragmenspárt ugyanannak a fehérjének egy másik röntgenszerkezetébe dokkoltam [T4].
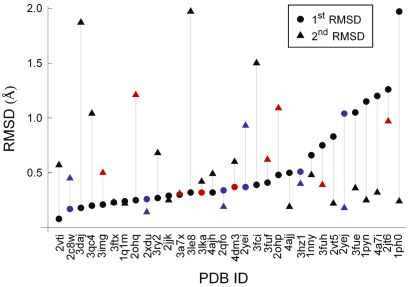
6. ábra. Két fragmens szekvenciális dokkolásának eredményei. A pontozási hibákat piros jelölők, a konzervált vízmolekulák jelenlétében végzett dokkolásokat kék jelölők mutatják.
Végül a szekvenciális dokkolási protokollt összekapcsolásra alkalmas fragmensek azonosítására használtam a D3 receptor kötőhelyén, és D2 homológia modellt is felhasználtam, mivel szelektív D3 antagonisták és parciális agonisták hatékonyak lehetnek skizofrénia, depresszió és bipoláris mánia kezelésében. Bázikus fragmensek dokkolásával a konzervált elsődleges kötőhelyre kötődő aril-piperazint azonosítottam. A másodlagos kötőhelyre dokkolt fragmensek közül egy ciklohexilamino-szulfonamid fragmens szelektivitásra utaló H-kötést alakított ki a Tyr1.39 aminosavval (7. ábra). Három összekapcsolt molekula előállítására került sor, és az ezt a fragmenst tartalmazó vegyület valóban magas affinitással és 55-szörös szelektivitással rendelkezett a D3 receptor javára [T5].
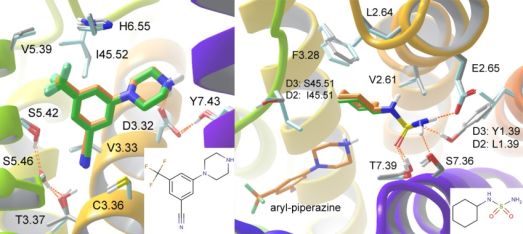
7. ábra. Az elsődleges (bal) és a másodlagos (jobb) kötőhelyre dokkolt fragmensek kötőmódjai. A D3 és D2 kötőhelyek szürke és világoskék színnel egymásra illesztve, a ligandumok dokkolt kötőmódjai a két kötőhelyen narancs és zöld színnel.
Várható impakt, további kutatás
A GPCR dúsulási tesztekről szóló publikációnk [T1] a 3. helyet érte el a JCIM legolvasottabb cikkei között, a prospektív GPCR fragmens szűrésről szóló cikket [T2] pedig a megjelenés utáni hónapban majdnem 500-an töltötték le, ami mutatja a cikkek iránti érdeklődést. Több fragmens dokkolásával kevesen foglalkoztak eddig [11], de a D3 fragmens kapcsolásról szóló cikk [T5] ACS Med Chem Lettersben való megjelenése szintén jelzi az alkalmazások iránti érdeklődést.
A bemutatott módszerek további GPCR-eken történő fragmens szűrésben alkalmazhatók, az azonosított D3 és H4 fragmensek pedig megfelelő kiindulási pontokként szolgálhatnak gyógyszerkémiai optimálás elindításához. A szekvenciális fragmensdokkolás szintén alkalmazható további célpontokkal szemben, továbbá a módszer segítheti a röntgenkrisztallográfiai szerkezetmegoldást ún. koktél fragmens-szűrés esetén [12].
Saját publikációk, hivatkozások, linkgyűjtemény
Kapcsolódó saját publikációk listája
T1. Tarcsay, Á., Paragi, G., Vass, M., Jójárt, B., Bogár, F., Keserű, G.M. The impact of molecular dynamics sampling on the performance of virtual screening against GPCRs. J. Chem. Inf. Model. 2013, 53, 2990-2999. (IF: 4.304)
T2. Vass, M., Schmidt, É., Horti, F., Keserű, G.M. Virtual fragment screening on GPCRs: a case study on dopamine D3 and histamine H4 receptors. Eur. J. Med. Chem. 2014, 77, 38-46. (IF: 3.499)
T3. Vass, M., Tarcsay, Á., Keserű, G.M. Multiple ligand docking by Glide: implications for virtual second-site screening. J. Comput. Aided Mol. Des. 2012, 26, 821-834. (IF: 3.172)
T4. Vass, M., Keserű, G.M. Fragments to link. A multiple docking strategy for second site binders. MedChemComm 2013, 4, 510-514. (IF: 2.722)
T5. Vass, M., Ágai-Csongor, É., Horti, F., Keserű, G.M. Multiple fragment docking and linking in primary and secondary pockets of dopamine receptors. ACS Med. Chem. Lett. Accepted. (IF: 3.311)
Hivatkozások listája
1. Leeson, P.D., St-Gallay, S.A. The influence of the 'organizational factor' on compound quality in drug discovery. Nat. Rev. Drug Discov. 2011, 10, 749.
2. Rees, D.C., Congreve, M., Murray, C.W., Carr, R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004, 3, 660.
3. Sheng, C., Zhang, W. Fragment informatics and computational fragment-based drug design: an overview and update. Med. Res. Rev. 2013, 33, 554.
4. Congreve, M., Carr, R., Murray, C., Jhoti, H. A 'rule of three' for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876.
5. Sándor, M., Kiss, R., Keserű, G.M. Virtual fragment docking by Glide: a validation study on 190 protein-fragment complexes. J. Chem. Inf. Model. 2010, 50, 1165.
6. de Graaf, C., Kooistra, A.J. et al. Crystal structure-based virtual screening for fragment-like ligands of the human histamine H1 receptor. J. Med. Chem. 2011, 54, 8195.
7. Chen, D., Ranganathan, A. et al. Complementarity between in silico and biophysical screening approaches in fragment-based lead discovery against the A2A adenosine receptor. J. Chem. Inf. Model. 2013, 53, 2701.
8. Tsai, J., Lee, J.T. et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041.
9. Ichihara, O., Barker, J., Law, R.J., Whittaker, M. Compound design by fragment-linking. Mol. Inf. 2011, 30, 298.
10. Hopkins, A.L., Keserű, G.M. et al. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105.
11. Hoffer, L., Horvath, D. S4MPLE - sampler for multiple protein-ligand entities: simultaneous docking of several entities. J. Chem. Inf. Model. 2013, 53, 88.
12. Nair, P.C., Malde, A.K., Drinkwater, N., Mark, A.E. Missing fragments: detecting cooperative binding in fragment-based drug design. ACS Med. Chem. Lett. 2012, 3, 322.
Linkgyűjtemény
Érdekességek a fragmensalapú gyógyszerkutatásból