 |
BMe Kutatói pályázat |

|

BME Oláh György Doktori Iskola
Richter Gedeon Nyrt.
Témavezető: Dr. Keserű György Miklós
Számításos modellek a gyógyszerkutatás támogatására
A kutatási téma néhány soros bemutatása
A gyógyszerkutatás alapvető célja olyan hatékony és biztonságos vegyületek előállítása, amelyek egy-egy adott betegség gyógyításán keresztül jelentősen hozzájárulnak az emberi életminőség javulásához. Az emberi szervezet sokrétű biokémiai folyamatai nagyfokú komplexitást mutatnak, ezért a gyógyszerkutatás egy sok dimenziós optimalizálási feladat. Munkám során számítási módszerek kidolgozását és jellemző összefüggések feltárását tűztem ki célul, amelyek segítik a gyógyszer-jelölt vegyületek hatékony és racionális tervezését.
A kutatóhely rövid bemutatása
A Richter Gedeon Nyrt. Közép-Európa egyik legnagyobb originális kutatóközpontjával rendelkezik. Az eredeti kutatás célja a központi idegrendszeri betegségek kismolekulás vegyületekkel történő kezelése. Az elmúlt évtizedek színvonalas munkáját a korábbi gyógyszerek (Cavinton és Toliperizone) mellett a jelenleg törzskönyvezés alatt álló Cariprazine jellemzi, amely skizofrénia és mánia indikációkban sikeres fázis III vizsgálatokkal rendelkezik.
A kutatás történetének, tágabb kontextusának bemutatása
A gyógyszer-jelölt vegyületekkel szemben támasztott követelmények nagyszámú, preklinikai fázisban mérhető paraméter eredőjeként kerülnek megfogalmazásra. A gyógyszer-jelöltek klinikai vizsgálatai során a hatékonyságot egyaránt befolyásolja a célfehérjével (célfehérjékkel) mutatott kölcsönhatás(ok) (affinitás, biokémiai funkcionális válasz, kötődési kinetika), valamint a farmakokinetikai tulajdonságok összessége (felszívódás, szervezeten belüli eloszlás, lebontás, kiürülés) lásd 1. ábra [1-2]. A biztonságosságot befolyásolja a mellékhatásokért felelős biokémiai folyamatok módosítása, mint az például élettani folyamatokban szereplő enzimek és receptorok működésének gátlása. A humán genomban jelen lévő több mint 30000 releváns gén [3] által kódolt fehérjék közül csupán egynek vagy néhánynak kell kölcsönhatásba lépnie a gyógyszerrel, Tell Vilmos próbatételéhez hasonlatos helyzetet teremtve a gyógyszerkutatók számára. A lehetséges mellékhatások között említhető a szívritmust befolyásoló hERG ioncsatorna gátlása (szívroham), a citokróm enzimrendszer működésének gátlása (májtoxicitás), illetve a sejtmembránnal való kölcsönhatás, amely sejtkárosodáshoz vezet (foszfolipidózis)[4].
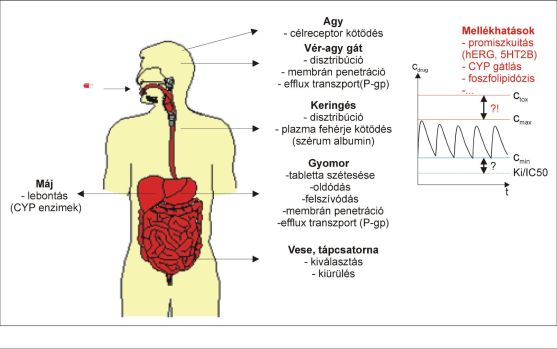
1. ábra. A gyógyszer sorsa a szervezetben.
A gyógyszertervezés feladatának szellemes hasonlata a Rubik kocka megoldása [5]. A 6 színből álló 54 négyzet ~43*1018 lehetséges kombinációt jelent (ez eltörpül a gyógyszerkémiai szempontból érdekes 1060 lehetséges vegyületszámhoz képest), azonban bármilyen kiindulópontból minimálisan 20 lépéssel megoldható. A gyógyszerkémiai optimalizálás során tipikusan ~100-3000 vegyületet állítanak elő célpontonként, azonban retrospektív elemzésekkel kimutatható, hogy a végtermék 20-60 lépésből levezethető a kiindulási szerkezetből, így a hatékony stratégia megtalálása kulcsfontosságú ( 2. ábra)[6].
A gyógyszertervezés első lépéseként a molekulatervezés számítási eszközök segítségével állít fel általános szabályokat (pl. korszakalkotó Lipinski-szabályok [7]), illetve fejleszt módszereket, amelyekkel a szintézist megelőzően becsülhetővé válnak a vegyület későbbi tulajdonságai (pl. affinitás, toxicitás).
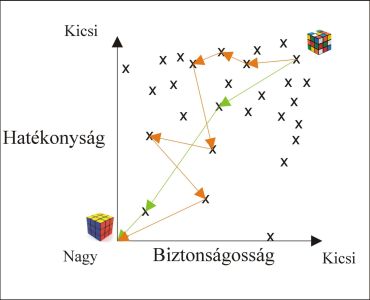
2. ábra A gyógyszerkémiai optimalizálás sematikus ábrája. A zöld útvonalon végrehajtott optimalizálás hatékonyabb, mint a narancssárga trajektória. A pontok hipotetikus vegyületeket jelölnek.
A kutatás célja, a megválaszolandó kérdések
Munkám során atomi szintű és holisztikus szemléletű problémák megfogalmazásával, két perspektívából végzetem vizsgálatokat. Elsőként a farmakokinetikai tulajdonságokat jelentősen meghatározó fehérjék közül a lebontásban szerepet játszó citokrómP450 enzimcsalád (CYP) vizsgálatát, valamint a felszívódásban és eloszlásban alapvető fontosságú P-glikoprotein (P-gp ) atomi szintű vizsgálatát tűztem ki célul.
Az alkalmazott molekulatervezési módszerek alapvetően két csoportba oszthatók. A fehérje térszerkezet ismeretében fehérje-ligandum kölcsönhatások modellezhetőek (szerkezet-alapú módszerek), illetve amennyiben az információk a ligandumokra korlátozódnak, ismert affinitású vegyületek térbeli illesztését végző, ligandum alapú módszerek használhatók. A röntgenkrisztallográfia dinamikus fejlődése az elmúlt évtizedben számos, gyógyszerkutatás szempontjából kiemelkedő jelentőségű eredményt szolgáltatott. 2003-ban közölték a humán CYP2C9 kristályszerkezetét [8], illetve 2009-ben az egér P-gp kristályszerkezetét [9], ezzel megteremtve a szerkezet-alapú módszerek alkalmazhatóságának feltételét.
Célom volt egy új szerkezet-alapú módszer kidolgozása és tesztelése a vegyületek metabolikusan instabil pontjának előrejelzésére.
Az egér P-glikoprotein fehérjeszerkezete alapján célul tűztem ki a humán enzim térszerkezetének becslését és a modell alkalmazhatóságának vizsgálatát.
Makroszkópikus megközelítéssel tanulmányoztam a vegyületek affinitásának és fizikokémiai jellemzőinek eloszlását, keresve az optimális tartományokat. Illetve célul tűztem ki a gyógyszerkémiai optimalizálás során alkalmazott lipofil hatékonyság mérőszámainak vizsgálatát. Relevanciájuk megértésére farmakokinetikai paraméterekkel való közvetlen összefüggéseket kerestem.
Módszerek
A fehérjék nagy atomszáma miatt a vizsgált rendszerek jellemzően molekulamechnikai (MM) módszerekkel vizsgálhatók. Munkám során kismolekulás ligandumok konformációkeresését végeztem a fehérje kötőhely által meghatározott térben (dokkolás). A dokkolás célja a kötőkonformáció és a kötődési szabadenergia becslése. A dokkolásokat Glide gyorsdokkoló programmal végeztem, amely irodalmi adatok alapján jó teljesítménnyel jellemezhető és a számítógépes gyógyszertervezés eszköztárának elfogadott eleme [10]. A dokkolás során a ligandum flexibilis, rigid fehérjeszerkezet mellett. A dokkolás egy speciális esete az indukált illeszkedés (IFD), amely nagyobb számítási kapacitás terhére figyelembe veszi a kötőhely flexibilitását is.
A homológiamodellezés során egy fehérje térszerkezetét ismert térszerkezetű, homológ szekvenciájú fehérje alapján becsüljük. A módszer alapját az elsődleges szerkezeti elemekben hasonlóságot mutató fehérjéknél tapasztalt hasonló harmadlagos szerkezet adja.
A ligandum célfehérjével alkotott komplexének jellemzésére leszorításos kísérletekből nyert Ki egyensúlyi állandó, vagy a biokémiailag mérhető jel 50%-os gátláshoz/indukálásához tartozó koncentráció (IC50/EC50) használható. A ligandum kötőhellyel való komplementaritását jól jellemzi az egy atomra eső kötődési szabadenergia változás, a ligandum-hatékonyság (LE).
A lipofilitás egy kulcsfontosságú fizikokémiai paraméter a gyógyszerkémiai optimalizálás során, amelyet tipikusan az oktanol-víz megoszlási hányados logaritmusával (logP), illetve a pH-függő logP, más néven logD értékkel adnak meg. A logP vagy logD értékek számos farmakokinetikai és toxikológiai paraméterrel mutatnak összefüggést [11-12]. A lipofil hatékonysági metrikák alapgondolata az affinitás és a lipofilitás egy mérőszámmal történő jellemzése. Leeson és Springthorpe definiálták az LLE=pAct-logP összefüggést, ahol pAct a Ki vagy IC50 tízes alapú negatív logaritmusa[13]. Keserű és Makara vezették be a LELP összefüggést, amely a ligandum-hatékonyság és a lipofilitás hányadosa: LELP=LE/logp [14]. Az LLE maximuma, míg a LELP minimuma jelent kedvezőbb lipofil hatékonyságot.
Eddigi eredmények
Metabolit szerkezet előrejelzés
A CYP450cam, a CYP enzimcsalád biokémiailag legjobban karakterizált izoformájának katalitikus ciklusát röntgenkrisztallográfiai szerkezetmeghatározással követve leírták, hogy a szubsztrát (kámfor), a reakció köztitermékei és a termék (hidroxi-kámfor) térbeli helyzete jelentős átfedést mutat [15] (3. ábra). Ez az izgalmas felfedezés vezetett a termék-dokkolás, vagy fordított-dokkolás módszerhez. Az eljárás azon alapul, hogy a termék illeszkedik az enzim aktív helyére így a biotranszformáció során beépített atomjának az enzim aktív centruma közelében kell lennie. A korábbi, szubsztrát-dokkoláson alapuló módszerekhez képes az új térbeli kényszerfeltétel többletinformációt jelent, amivel pontosítható a konformációkeresés eredménye (4. ábra)[T1].
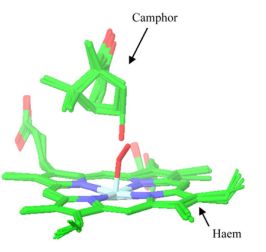
3. ábra. P450cam enzim katalitikus ciklusa során nyert röntgenszerkezetek illesztése kámfor ligandummal[15].
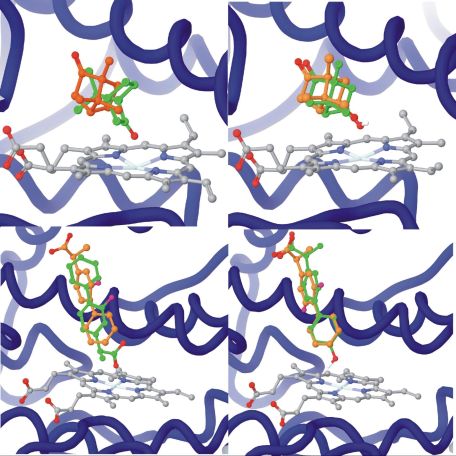
4. ábra. Szubsztrát-dokkolás és termék-dokkolás összehasonlítása[T1]. A felső sorban a kámfor (P450cam), alsó sorban a flurbiprofen kötőmódja, szubsztrát- (bal oldal), illetve termék- (jobb oldal) dokkolás esetén. Mindkét molekula esetén a jobb oldali oszlop mutat nagyobb átfedést a kristályszerkezetben található (zöld) és dokkolással szimulált (narancssárga) megoldások között.
A módszer három lépésből tevődik össze: 1) lehetséges termékek enumerálása, 2) kötőkonformációk előállítása és szűrése távolság kényszer alapján, 3) lehetséges termékek rangsorolása az enzimmel való komplementaritás alapján.
A módszer teljesítőképességét a humán CYP2C9 enzimen vizsgáltam. A dokkolás során első három helyen rangsorolt lehetséges termékek az esetek 83,7%-ában tartalmazták a valósat, felülmúlva a szubsztrát dokkolás eredményét (47%). Hatékonysága az irodalomban megtalálható, más elven működő alkalmazások hatékonyságához hasonló [T2]. Előnye, hogy a metabolikus hely előrejelzésén túl megadja a termék szerkezetét, továbbá adaptációja a dokkoló programon kívül nem igényli különleges célprogram használatát (a lehetséges termékek elvileg bármilyen enumerálással előállíthatók).
A CYP enzimekre jellemző a kooperatív kötődés, amelyet kísérletileg a CYP3A4 izoforma ketokonazollal kristályosított szerkezete is bizonyít: a ligandum két kópiában található a kötőhelyen. Munkatársaimmal kifejlesztettünk egy szekvenciális eljárást a kooperatív ligandumok kötőmódjának előrejelzésére. Ez a módszer segítséget nyújthat CYP enzimek esetén a többszörös kötődés atomi szintű megértésében[T3].
P-gp homológiamodell építés
Az egér P-gp kristályszerkezete alapján modelleztem a humán ortológ térszerkezetét [T4]. A homológiamodellt sztereokémiai (Ramachandran-eloszlás) és energetikai (ProSa) szempontok alapján validáltam. Négy, biokémiai módszerekkel behatóan tanulmányozott ligandum indukált illeszkedéssel szimulált kötőmódjának elemzése során jó egyezést találtam az indirekt biokémiai eredményekkel. Merev fehérje-dokkolással vizsgáltam P-gp ligandumok virtuális szűréssel történő rangsorolásának lehetőségét. A merev fehérjemodell alkalmazásával kapott gyenge eredmények és az indukált illeszkedéssel kapott kötőmódok elemzése alapján megállapítottam, hogy a P-gp esetén kiemelkedő fontosságú a fehérje mozgékonyságának figyelembevétele, ezért gyorsdokkoló eljárásokkal történő virtuális szűrésre nem alkalmas.
Szelektivitás
A gyógyszerek mellékhatásprofilját jelentősen meghatározza a célfehérjén kívül más fehérjékkel mutatott kölcsönhatási mintázata (promiszkuitás). Az irodalmi adatok összefoglalása megmutatta, hogy a lipofilitás és a vegyületek bázikus karaktere jelentős összefüggést mutat a promiszkuitással (5. ábra) [T5]. A vizsgált adatkészleten a kevésbé lipofil (logP<4) vegyületek szignifikánsan kisebb promiszkuitást mutattak, mint az ennél magasabb logP értékkel rendelkező vegyületcsoport (Student t-próba, p<0,001).
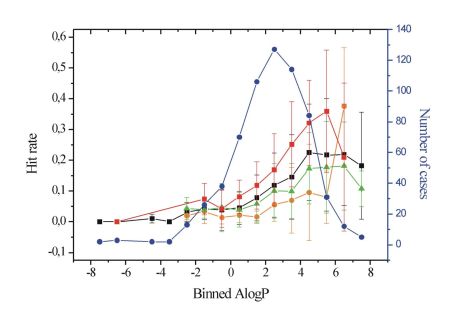
5. ábra. Lipofilitás (AlogP) és nagyszámú célfehérjén mért találati arány (Hit rate) összefüggése[T5].
A promiszkuitás elkerülésére javaslatot tettem az alábbi ábrán látható működési ciklus széleskörű használatára a gyógyszerkémiai programokban (6. ábra). Az ionizáció fontossága kapcsán, mint alkalmazott számított előszűrő, az előrejelzés pontosságát egy összehasonlító tanulmányban vizsgáltam[T6].
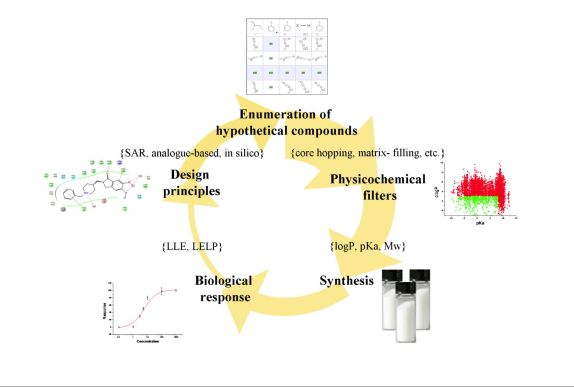
6. ábra. Javasolt gyógyszerkémiai optimalizációs ciklus[T5].
Lipofil hatékonyság
A preklinikai gyógyszerkutatás életciklusának (találatvegyület, vezérmolekula, gyógyszer-kandidátus, klinikai vizsgálatok) függvényében vizsgáltam két lipofil hatékonysági metrika az LLE és a LELP értékeinek eloszlását (7. ábra)[T7].
Eredményeim alátámasztották ezen metrikák használatának fontosságát, hiszen a gyógyszerek és a sikeres fázis I. vizsgálattal rendelkező vegyületek elkülönültek a korábbi fázisokban található vegyületektől. Így hatékony stratégia lehet a lipofil hatékonysági metrikák értékeit a korai optimalizálásban a sikeres vegyületeknek megfelelő tartománya felé változtatni. A LELP esetén magasabb korrelációt találtam hat farmakokinetikai mérési eredményen elért előnyös tulajdonságok és a kedvezőbb LELP értékek között (8. ábra). Munkám során különböző szubsztitúciók LELP változásait elemezve gyakorlatban alkalmazható módosítási sémát vetettem fel.
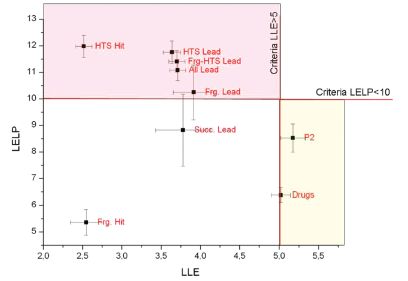
7. ábra. LLE és LELP átlagértéke és szórása[T8]. Hit: találatmolekula, Lead: vezérmolekula, Frg: fragmens optimalizálás, HTS: nagy áteresztőképességű szűrés , P2: humán klinikai vizsgálat II. fázisban lévő vegyületek, Drugs: gyógyszerek.
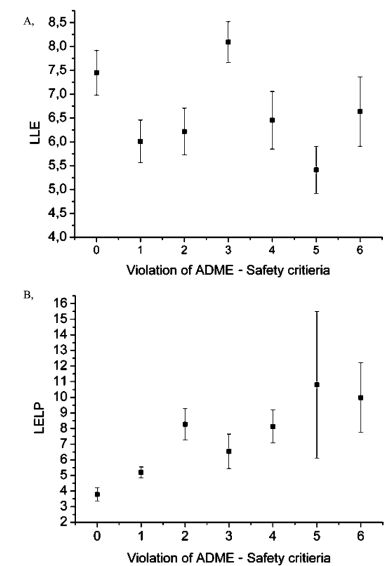
8. ábra. LLE és LELP átlagértékek változása hat farmakokinetikai és toxikológiai mérésen elér előnytelen értékek (Violation of ADME-Safety criteria) függvényében[T7].
Várható impakt, további kutatás
A visszajelzések alapján eredményeimet a szakmai közvélemény érdeklődéssel fogadta. Két cikk, [T5] és [T7], szerepelt a Journal of Medicinal Chemistry 20 legolvasottabb cikke között az 1 hónapos, illetve 1 éves felbontásban készült statisztikák alapján, valamint a megjelenés évében az első negyedév 6 legolvasottabb cikke között. A lipofil hatékonysággal kapcsolatos közleményt [T7] John Lowe értékelte és ajánlotta F1000Prime válogatásban[16], így bekerült a biológia és gyógyszerkémia témakörben megjelent publikációk rangsorának felső 2%-ába.
2012-es megjelenése óta 20 cikkben hivatkozzák.
A pályázatban hivatkozott saját publikációkra több mint 60 hivatkozás történt.
A jövőben Freire és munkatársainak hipotézise [17] alapján a kötődési termodinamikai paramétereknek, vagyis a fehérje-ligandum kölcsönhatás minőségének, a szelektivitással való összefüggésének vizsgálatát tervezem.
Saját publikációk, hivatkozások, linkgyűjtemény
Kapcsolódó saját publikációk listája
[T1] Tarcsay, A.; Kiss, R., Keserű, G.M. Site of metabolism prediction on cytochrome P450 2C9: a knowledge-based docking approach. J Comput Aided Mol Des 2010, 24,399-408. old. (IF:3.386)
[T2] Tarcsay, Á., Keserű, G.M. In silico site of metabolism prediction of cytochrome P450-mediated biotransformations. Expert Opin Drug Metab Toxicol. 2011, 7, 299-312. old. (IF:3.119)
[T3] Vass, M.; Tarcsay, Á., Keserű, G.M. Multiple ligand docking by Glide: implications for virtual second-site screening. J Comput Aided Mol Des. 2012, 26, 821-34. old. (IF:3.386)
[T4] Tarcsay, A., Keserű, G.M. Homology modeling and binding site assessment of the human P-glycoprotein. Future Med Chem. 2011, 3, 297-307. old. (IF:2.522)
[T5] Tarcsay, Á., Keserű G.M. Contributions of molecular properties to drug promiscuity.
J Med Chem. 2013, 56, 1789-1795. old. (IF: 5.248)
[T6] Balogh, G.T., Tarcsay, A.; Keserű, G.M. Comparative evaluation of pK(a) prediction tools on a drug discovery dataset. J Pharm Biomed Anal. 2012 67-68, 63-70. old. (IF:2.967)
[T7] Tarcsay, A.; Nyíri, K.; Keserű, G.M., Impact of lipophilic efficiency on compound quality. J Med Chem. 2012, 55, 1252-1260. old. (IF: 5.248)
Linkgyűjtemény
Hivatkozások listája
[1] van de Waterbeemd, H., Gifford, E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov. 2003, 2(3), 192-204. old.
[2] Pan, A.C.; Borhani, D.W.; Dror, R.O., Shaw, D.E.Molecular determinants of drug-receptor binding kinetics. Drug Discov Today. 2013 Feb 27. pii: S1359-6446(13)00062-7. doi: 10.1016/j.drudis.2013.02.007. [Epub ahead of print]
[3] Hopkins, A.L., Groom, C.R. The druggable genome. Nat Rev Drug Discov. 2002, 1(9), 727-730. old.
[4] Antitagets. Roy J Vaz, Thomas Klaubinde (Szerk.), Wiley, 2008, Weinheim.
[5] Lusher, S.J.; McGuire, R.; Azevedo, R.; Boiten, J.W.; van Schaik, R.C.; de Vlieg, J., A molecular informatics view on best practice in multi-parameter compound optimization. Drug Discov Today. 2011, 16, 555-68. old.
[6] Cheshire, D.R., How well do medicinal chemists learn from experience? 2011, Drug Discov Today. , 16, 817-21. old.
[7] Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J., Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001, 46 (1-3), 3-26. old.
[8] Williams, P. A.; Cosme, J.; Ward, A.; et al., Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003, 424, 464-8. old.
[9] Aller, S.G.; Yu, J.; Ward, A. et al., Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science, 2009, 323(5922), 1718–1722. old.
[10] McGaughey, G.B.; Sheridan, R.P.; Bayly, C.I.; Culberson, J.C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, J.F.; Cornell, W.D., Comparison of topological, shape, and docking methods in virtual screening. J Chem Inf Model, 2007, 47, 1504–1519. old.
[11] Gleeson, M.P., Generation of a set of simple, interpretable ADMET rules of thumb.J Med Chem. 2008, 51 (4), 817-834. old.
[12] Waring, M.J., Lipophilicity in drug discovery. Expert Opin Drug Discov. 2010, 5 (3), 235-48. old.
[13] Leeson, P. D.; Springthorpe, B., The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6:881-890. old.
[14] Keserű, G. M.; Makara, G. M., The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discovery 2009, 8:203-212. old.
[15] Schlichting, I.; Berendzen, J.; Chu. K. et al., The catalytic pathway of cytochrome p450cam at atomic resolution. Science 2000, 287, 1615-1622. old.
[16] Lowe J., F1000Prime Recommendation of [Tarcsay A et al., J Med Chem 2012, 55(3):1252-60]. In F1000Prime, 24 Jan 2012; DOI: 10.3410/f.13484092.14861219. F1000Prime.com/13484092#eval14861219
[17] Kawasaki, Y.; Freire, E. Finding a better path to drug selectivity. Drug Discov Today. 2011, 16, 985-990. old.