 |
BMe Kutatói pályázat |

|

Oláh György Doktori Iskola
BME VBK, Szervetlen és Analitikai Kémia Tanszék
Témavezető: Dr. Szieberth Dénes
Hidrogénaktiválás nemfémes elemekkel
A kutatási téma néhány soros bemutatása
Ipari és laboratóriumi gyakorlatban sokszor előfordul, hogy telítetlen, kettős, illetve hármas kötéseket tartalmazó molekulákat szeretnénk telíteni például hidrogénnel. A hidrogénmolekulában lévő kötést igen nehéz felbontani, ezért a hidrogénmolekula aktiválása (reakcióképességének növelése) rendkívüli fontossággal bíró feladat a kémiában: például petrolkémia, élelmiszerkémia, gyógyszerkémia, hidrogéntárolás területén[1]. A hidrogén katalitikus bontásához általában átmenetifémeket használnak katalizátorként, manapság azonban előtérbe került a nemfémes katalizátorok használata is, mivel egyes átmenetifémek nagyon drágák, illetve környezeti szempontból károsak lehetnek. Doktori munkám során számításos kémiai módszerek segítségével ilyen nemfémes, hidrogén aktiválására alkalmas katalizátorok fejlesztésével foglalkozom.
A kutatóhely rövid bemutatása
A kutatást a BME-VBK Szervetlen és Analitikai Kémia Tanszéken folytatom Dr. Szieberth Dénes témavezetésével. A tanszéken Dr. Nyulászi László és csoportja már régóta foglalkozik elméleti kémiával, azon belül ionos folyadékok, karbének, főcsoportbeli elemek kémiájával. A csoport eddigi kutatási eredményeiből számos nemzetközileg elismert publikáció született.

Elméleti Kémia Csoport (BME-VBK, SzAKT)
A kutatás történetének, tágabb kontextusának bemutatása
Manapság a nemfémes katalizátorok használata hidrogénaktiválási reakciókban intenzíven kutatott terület [2,3,4]. Ilyen organokatalizátorok tervezésekor gyakran kihasználják a frusztrált Lewis sav-bázis pár (FLP) elméletet[5]. A frusztrált Lewis-pár általában egy Lewis-sav (elektron akceptor) és egy Lewis-bázis (elektrondonor) által alkotott molekulapár, melyek a rajtuk lévő nagy térkitöltésű csoportok akadályozó hatása miatt nem tudnak egymással datív kötést kialakítani. A molekulapárban így kialakuló reaktív centrum képes kis molekulákat, így például a hidrogént is aktiválni (1. ábra), sőt egyes esetekben ez a folyamat reverzibilis[5,6]. Egy hidrogénező katalizátornál a reverzibilitás azért fontos, mert amellett, hogy a katalizátor aktiválja a hidrogént, el is kell tudnia engedni azt; nem keletkezhet túlzottan stabil hidrogén addukt.
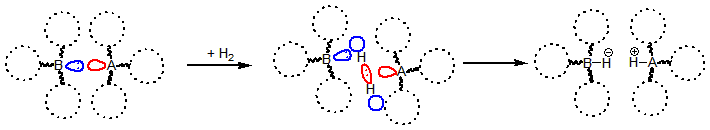
1. ábra: Frusztrált Lewis-párokkal történő hidrogénaktiválás
A hidrogénaktiválási mechanizmus a frusztrált Lewis-pároknál más, mint az átmenetifémeknél (2. ábra). Az átmenetifémek általában egy köztitermék (intermedier) keletkezése közben, kétlépéses reakcióban aktiválják a hidrogént[7].
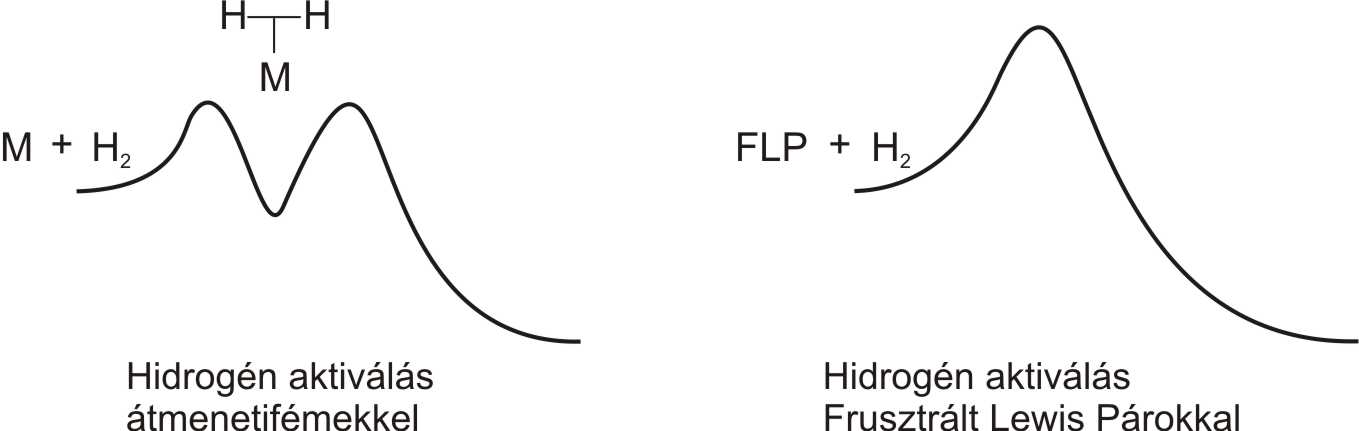
2. ábra: A hidrogénaktiválás lehetséges mechanizmusai (bal: átmenetifémekkel, jobb: frusztrált Lewis-párokkal)
A reakció során keletkező η2-H2 komplex intermediert két jelentős kölcsönhatás stabilizálja: egyrészt a H2 molekula σ-kötése elektront donál a fém egyik üres d-pályájára, továbbá a fém d-típusú nemkötő elektronpárja elektront (vissza)donál a H2 σ*-lazítópályájára[7]. E két kölcsönhatás eredményeként a H−H kötés megnyúlik, a hidrogénmolekula aktiválódik. A 3. ábrán látható egy példa, ahol egy központi krómatom komplexál két CO molekulát, egy benzolt és egy H2 molekulát[8].
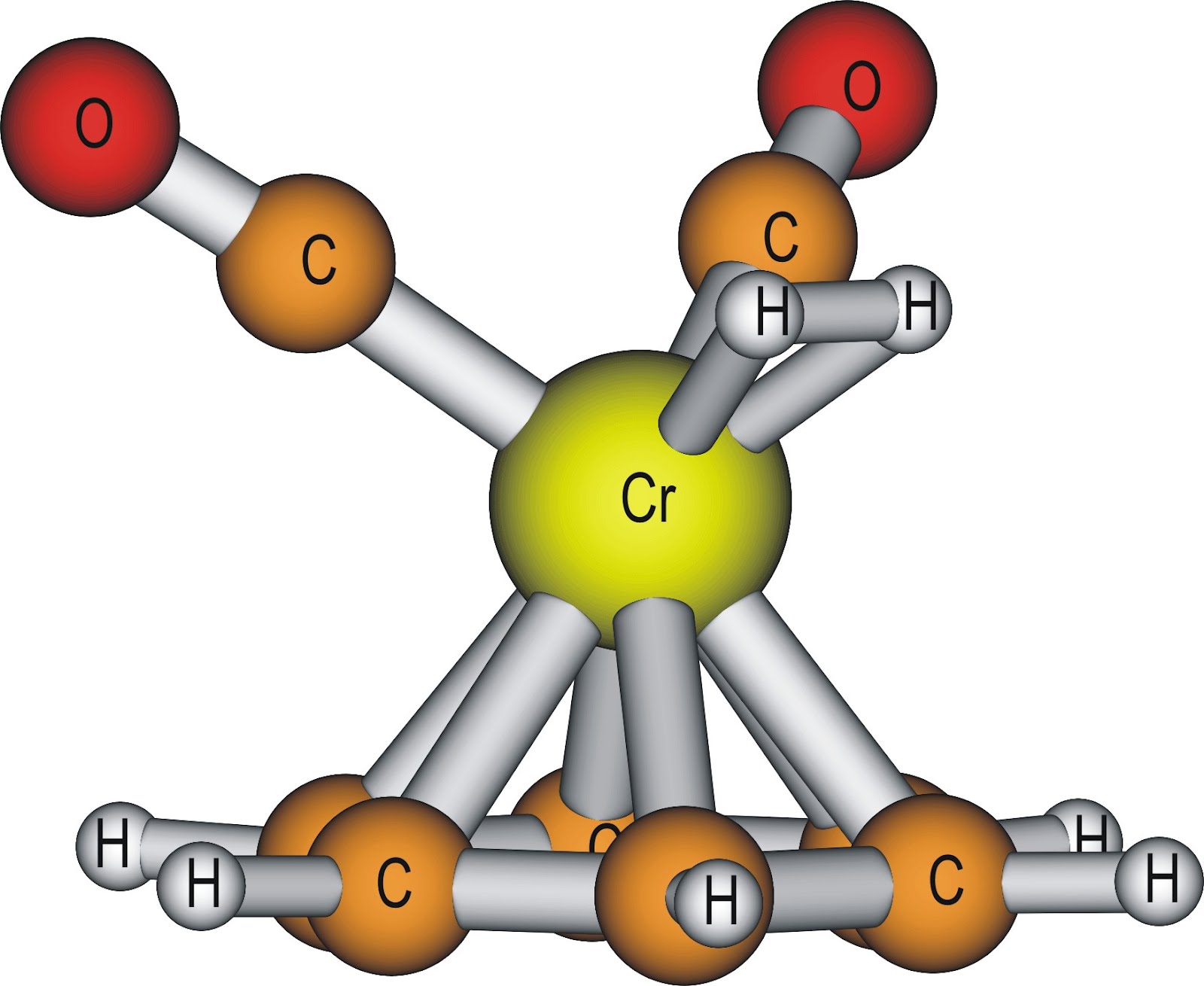
3. ábra: (η6-benzol)(CO)2Cr(η2-H2) komplex
A kutatás célja, a megválaszolandó kérdések
A frusztrált sav-bázis párok teljesen más mechanizmusú egylépéses reakcióban aktiválják a hidrogént, amely során nem található az átmenetifémekre jellemző η2-H2 komplex a reakcióúton [9]. Az átmeneti fémekkel ellentétben itt az elektron akceptor és az elektrondonor nem egy centrumon (a fémen) lokalizálódik, hanem kettőn. E két centrum (Lewis-sav és a Lewis-bázis) erős polarizációs hatás következtében azonnal bontja a H−H kötést, és nem tud kialakulni stabil η2 komplex [9]. Az általunk vizsgált difoszfino-borán egy olyan FLP, amely esetén mégis kialakul egy stabil intermedier (lásd később: Eddigi eredményeink fejezet). Így lehetőség nyílik egy olyan reakcióút megvalósulására, amikor egy nemfémes molekula utánozza az átmenetifémekre jellemző hidrogénaktiválási mechanizmust és köztiterméket (4. ábra).
4. ábra: Difoszfino-boránok η2-H2 komplexe
A tervezett kutatás egyik célja annak vizsgálata, hogy miként befolyásolják különböző típusú helyettesítők az η2-H2 difoszfino-borán komplex stabilitását és a hidrogénaktiválás reakciógátját. Ezzel párhuzamosan szeretnénk megvizsgálni, hogy miként stabilizálhatók az elektrondonor csoportokkal nem rendelkező η2-H2 BR3 komplexek. Az irodalomban eddig csak néhány példa van η2-H2 borán komplexre, és ezek közül is csak a BH5-öt sikerült előállítani − kripton mátrixban [10]. Számítások szerint minden esetben, ha egy külső bázist adunk az ilyen típusú η2-H2 borán komplexekhez, a H−H kötés rögtön polarizálódik, és azonnal bomlik [9].
Módszerek
A kutatás során különböző ab initio számításos kémiai módszerek és azokat implementáló kvantumkémiai szoftverek segítségével szeretnénk válaszolni a fent említett kérdésekre. Elméleti kémiai módszerek segítségével könnyen lehet modellezni molekulák szerkezetét, reakciók mechanizmusát. Egy adott komplex stabilitása is számos módon jellemezhető: a kötéstávolságok analízise és a kötésbontási reakciók energetikája mellett az elektronsűrűség topológiai analízise (AIM), illetve a természetes kötőpályák vizsgálata (NBO) is adatokat szolgáltat a stabilitás becsléséhez. Az η2-H2 komplexek minél teljesebb jellemzésére e módszerek együttesét szeretnénk használni.
A kvantumkémiai modellezést minden esetben meg kell előznie előzetes vizsgálatoknak, melyek során ki kell választani a megfelelő számítási szintet (módszer és bázis készlet), amely jól leírja az adott problémát. Ezért a kutatás első felében tesztszámításokat kell végezni, mellyel ellenőrizzük a választott számítási szint jóságát. A vizsgálandó molekulák mérete miatt előreláthatólag a sűrűségfunkcionál elmélet (DFT) fogja adni a megfelelő kompromisszumot a számítás pontossága és gépigénye között. A probléma leírásához megfelelő DFT funkcionál kiválasztását kisebb tesztmolekulákra végzett magasabb szintű ab initio (MP2, CCSD(T)) számításokkal fogjuk végezni. Ezután lehet hozzákezdeni az adott probléma részletes tanulmányozásához.
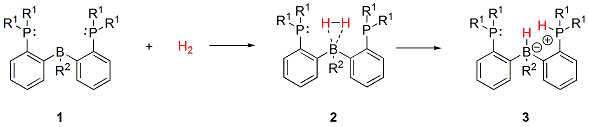
5. ábra: Difoszfino-boránok reverzibilis hidrogénaktiválási reakciója
R1: −CH3, −iPr, −tBu, −SiH3, −NiPr2 ; R2: −H, −CH3, −Ph, −Cl, −CF3, −SiH3, −SiMe3
Korábbi számítások azt sugallják, hogy a difoszfino-borán komplex stabilizálásához komplexben levő foszfornak erősíteni kell az elektrondonor, a bórnak pedig az elektron akceptor képességét[11]. Éppen ezért a következő szubsztituensekkel (például nagy térkitöltésű sztérikus csoportokkal és/vagy elektronszívó, elektronküldő csoportokkal) szeretnénk kiszámolni az 5. ábrán látható egyenlet reakcióprofilját: R1: −CH3, −iPr, −tBu, −SiH3, −NiPr2 ; R2: −H, −CH3, −Ph, −Cl, −CF3, −SiH3, −SiMe3. Vizsgálnánk továbbá azt is, hogy mi történik, ha kicseréljük a központi atomokat más Lewis-bázisra, illetve savra (például bór helyett alumínium, foszforok helyett nitrogének).
Az elektrondonor csoportokkal nem rendelkező η2-H2 BR3 komplex stabilitásának vizsgálatához különböző sztérikus és elektronikus tulajdonságokkal rendelkező R helyettesítőkkel szeretnénk kiszámolni a következő reakciót: BR3 + H2 = BR3H2. A BR3H2 összegképletű η2-H2 borán komplexek (ha egyáltalán léteznek) könnyen elbomlanak, e molekulák stabilitását jól jellemzi a fentebb említett reakció reakcióhője. E komplexek instabilitását azzal magyarázzák, hogy a bór nem képes elektront (vissza)donálni a H2 σ*-lazítópályájára. Felvetődik azonban az a kérdés, hogy a komplex stabilizálásához szükséges (vissza)donálás nemcsak egy külső donorcsoportról, hanem a bór és a körülötte lévő R helyettesítők B−R kötéséből is származhat, így a polarizációs hatás és ezzel együtt a komplex széteséséhez vezető hajlam csökkenhet.
Eddigi eredmények
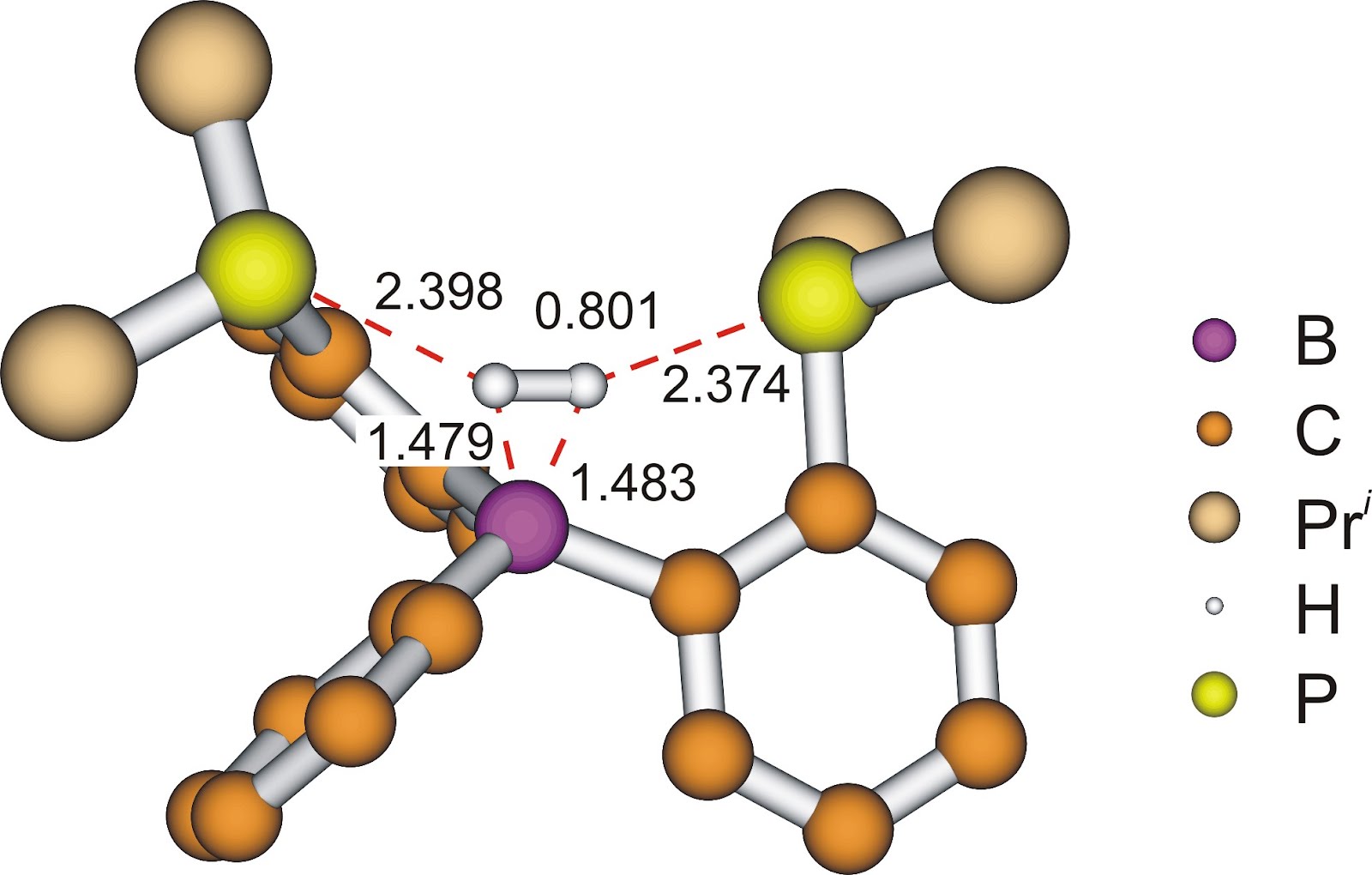
Eddigi munkánk során kimutattuk, hogy a difoszfino-boránok képesek a hidrogént reverzibilisen aktiválni[11] (5. ábra). A difoszfino-borán olyan frusztrált Lewis-pár, amelynek egy Lewis-sav (elektronpár-akceptor) és két Lewis-bázis (elektronpár-donor) funkciós csoportja van[12]. E csoportok szimmetrikus elrendeződése következtében a molekula képes imitálni az átmenetifémek η2-H2 komplexét (4. ábra v. 5. ábra: 2 molekula). Elméleti kémiai számítások szerint a komplexeket két fő kölcsönhatás stabilizálja. Egyrészt a két foszforatom nemkötő elektronpárjainak szimmetrikus elektrondonálása a hidrogénmolekula mindkét hidrogénatomja felé. Másrészt a hidrogénmolekula a rajta keletkező elektrontöbbletet továbbítja a bóratomra, ezzel erősítve a B−H2 kötést[11]. Ez az elektrontranszfer nyomon követhető a komplex elektronsűrűségének topológiai analízisével. A molekula elektronsűrűségéről háromdimenziós térkép készíthető, ezen a kémiai kötéseket a maximális elektronsűrűség által kijelölt kötésösvény, illetve az ezen kialakuló kötés kritikus pont jellemzi. A 6. ábrán a foszforatomok és a hidrogénmolekula, illetve hidrogénmolekula és bór között kialakuló kötésösvények láthatóak. A számítások igazolják azt is, hogy a köztitermék stabilitása jelentősen befolyásolja a hidrogénaktiválás sebességmeghatározó lépésének gátmagasságát.
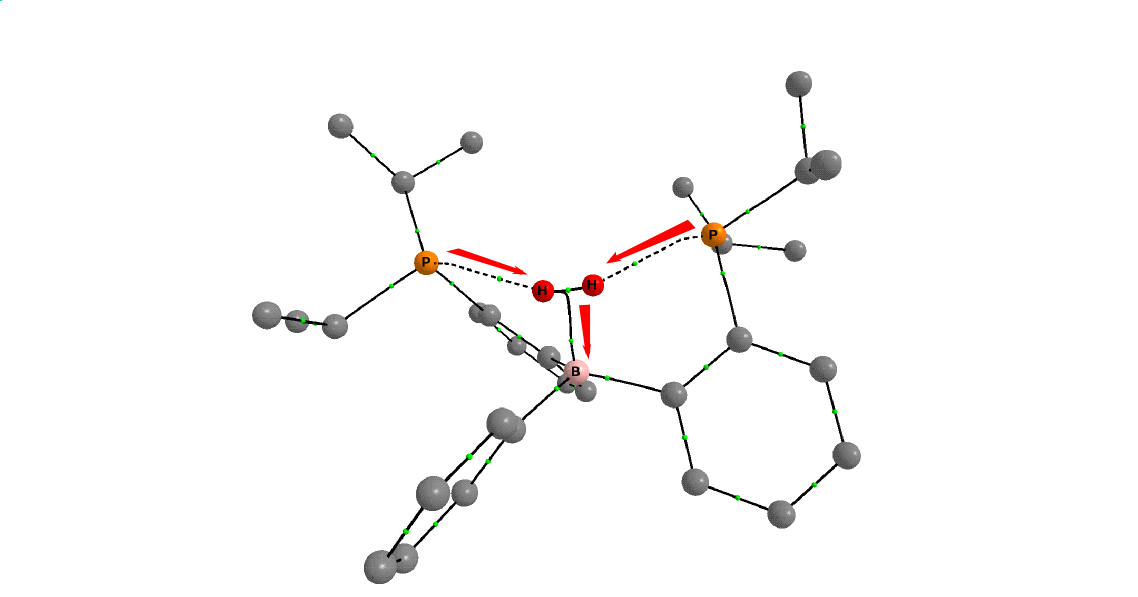
6. ábra: Difoszfino-boránok η2-H2 komplexének AIM ábrája
Várható impakt, további kutatás
A számítási eredmények alapján várhatóan egy új nemfémes hidrogénaktiválási mechanizmust térképezünk fel, ami új gondolkodásmódot vezethet be az FLP kémiába. Az egy molekulán belüli több donorcsoport alkalmazása új típusú katalizátorok fejlesztését teszi lehetővé, amelyekkel csökkenthető a hidrogénaktiválási reakciók gátja. A lehetséges célvegyületeket az együttműködő francia kutatócsoport segítségével szeretnénk szintetizálni. Továbbá eredményeink alapján esetleg meg tudunk jósolni stabil, előállítható η2-H2 borán komplexeket, amely komplexek igen érdekesek, hiszen ezekben a molekulákban a bór öt vegyértékű, és erre az irodalomban nagyon kevés példa van. Továbbiakban tervezzük ezen ötértékű bórvegyületek előállítását itt a tanszéken.
Saját publikációk, hivatkozások, linkgyűjtemény
Kapcsolódó saját publikációk listája:
L. Könczöl, A. Kawachi, D. Szieberth, Organometallics 2012, 31, 120-125
L. Könczöl, E. Makkos, D. Bourissou, D.Szieberth, Angew.Chem Int. Ed. elküldve
Linkgyűjtemény:
Hivatkozások listája:
[1] J. F. Hartwig, in Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA 2010
[2] a) A. L. Kenward, W. E. Piers, Angew. Chem. 2008, 120, 38–42; Angew. Chem. Int. Ed. 2008, 47, 38–41. b) D. W. Stephan, Chem. Commun. 2010, 46, 8526–8533. c) P. P. Power, Nature 2010, 463, 171–177. d) P. P. Power, Acc. Chem. Res. 2011, 44, 627–637.
[3] a) G. H. Spikes, J. C. Fettinger, P. P. Power, J. Am. Chem. Soc. 2005, 127, 12232–12233. b) Y. Peng, M. Brynda, B. D. Ellis, J. C. Fettinger, E. Rivard, P. P. Power Chem. Commun. 2008, 6042–6044. c) Y. Peng, B. D. Ellis, X. Wang, P. P. Power, J. Am. Chem. Soc. 2008, 130, 12268–12269. d) Y. Peng, J.-D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase, P. P. Power, J. Am. Chem. Soc. 2009, 131, 16272–16282. e) Z. Zhu, X. Wang, Y. Peng, H. Lei, J. C. Fettinger, E. Rivard, P. P. Power, Angew. Chem. 2009, 121, 2065–2068; Angew. Chem. Int. Ed. 2009, 48, 2031–2034.
[4] a) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller, G. Bertrand, Science 2007, 316, 439–441. b) D. Martin, M. Soleilhavoup, G. Bertrand, Chem. Sci. 2011, 2, 389–399.
[5] a) G. C. Welch, R. R. San Juan, J. D. Masuda, D. W. Stephan, Science 2006, 314, 1124–1126. b) P. A. Chase, G. C. Welch, T. Jurca, D. W. Stephan, Angew. Chem. 2007, 119, 8196–8199; Angew. Chem. Int. Ed. 2007, 46, 8050–8053. c) G. C. Welch D. W. Stephan, J. Am. Chem. Soc. 2007, 129, 1880–1881. d) P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme, D. W. Stephan, Chem. Commun. 2007, 5072–5074. e) P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Fröhlich, G. Erker, Angew. Chem. 2008, 120, 7654–7657; Angew. Chem. Int. Ed. 2008, 47, 7543–7546.
[6] D. W. Stephan, G. Erker, Angew. Chem. 2010, 122, 50–81; Angew. Chem. Int. Ed. 2010, 49, 46–76.
[7] a) R. H. Crabtree, Angew. Chem. 1993, 105, 828–845; Angew. Chem. Int. Ed. 1993, 32, 789–805. b) G. J. Kubas, in Metal dihydrogen and sigma–bond complexes; Kluwer Academic/Plenum ed.; New York, 2001. c) S. Aldridge, A. J. Downs, Chem. Rev. 2001, 101, 3305–3365. d) G. J. Kubas, Chem. Rev. 2007, 107, 4152–4205. e) R. D. Adams, B. Captain, Angew. Chem. 2008, 120, 258–263; Angew. Chem. Int. Ed. 2008, 47, 252–257. f) G. J. Kubas, J. Organomet. Chem. 2009, 694, 2648–2653.
[8] J. D. Egbert, D. M. Heinekey, Organometallics 2010, 29, 3387–3391.
[9] a) T. A. Rokob, A. Hamza, A. Stirling, T. Soós, I. Papai, Angew. Chem. 2008, 120, 2469–2472; Angew. Chem. Int. Ed. 2008, 47, 2435–2438. b) R. Rajeev, R. B. Sunoj, Chem. Eur. J. 2009, 15, 12846–12855. c) S. Grimme, H. Kruse, L. Goerigk, G. Erker, Angew. Chem. 2010, 122, 1444–1447; Angew. Chem. Int. Ed. 2010, 49, 1402–1405.
[10] a) T. J. Tague Jr., L. Andrews, J. Am. Chem. Soc. 1994, 116, 4970–4976. b) P. R. Schreiner, H. F. Schaefer III, P. v. R. Schleyer, J. Chem. Phys. 1994, 101, 7625–7632. c) J. D. Watts, R. J. Bartlett, J. Am. Chem. Soc. 1995, 117, 825–826. d) S. Fau, G. Frenking, Mol. Phys. 1999, 96, 519–527. e) O. A. Filippov, A. M. Filin, V. N. Tsupreva, N. V. Belkova, A. Lledós, G. Ujaque, L. M. Epstein, E. S. Shubina, Inorg. Chem. 2006, 45, 3086–3096. f) C. Fan, L. G. Mercier, W. E. Piers, H. M. Tuononen, M. Parvez, J. Am. Chem. Soc. 2010, 132, 9604–9606. g) Z. Lu, Z. Cheng, Z. Chen, L. Weng, Z. H. Li, H. Wang, Angew. Chem. 2011, 123, 12435–12439; Angew. Chem. Int. Ed. 2011, 50, 12227–12231. h) G. I. Nikonov, S. F. Vyboishchikov, O. G. Shirobokov, J. Am. Chem. Soc. 2012, 134, 5488–5491. d) J.-L. M. Abboud, B. Németh, J.-C. Guillemin, P.Burk, A. Adamson, E. R. Nerut, Chem. Eur. J. 2012, 18, 3981–3991.
[11] L. Könczöl, E. Makkos, D. Bourissou, D.Szieberth, Angew.Chem Int. Ed. elküldve
[12] S. Bontemps, G. Bouhadir, P. W. Dyer, K. Miqueu, D. Bourissou, Inorg. Chem. 2007, 46, 5149–5151.